Introduction
Microcystic stromal tumor (MCST) is a rare subtype of ovarian stromal tumor first described in 2009 by Irving et al. [1]. Histologically, MCST shows microcystic, solid cellular regions and a hyalinized fibrous stroma, with immunohistological features of positive staining for vimentin, CD10, β-catenin (nuclear location), and cyclin D1 [2]. MCSTs are presently presumed to be ovarian stromal in origin, though this hypothesis has not been firmly established. As a new entity within the category of sex cord-stromal neoplasms, the current 31 MCST cases reported in literature were found in the ovaries. In the male reproductive system, common sexcord stromal tumors include Leydig cell tumors, Sertoli cell tumors, and granulosa cell tumors, and tumors of the fibroma-thecoma group, which are quite rare. Moreover, there was no equivalent entity for MCST in the male reproductive system prior to this study. In this study, we present a unique case of a testicular tumor with a characteristic triad of cellular islands, microcystic spaces, and hyalinized bands, with a typical immuno-profile (CD10-positive, vimentin-positive, cyclin D1‒positive, β-catenin‒positive, and epithelial membrane antigen‒negative), as well as the absence of morphologic features that would indicate any other specific tumors in the sex cord-stromal category of the testis [3]. However, these features are consistent with MCST of the ovary. To the best of our knowledge, this was the first case of MCST of the testicle. The study was approved by the Institutional Review Board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (IRB No. TJ-C2016-0601) and performed in accordance with the principles of the Declaration of Helsinki. Written informed consents were obtained from the partient for this study.
Case Report
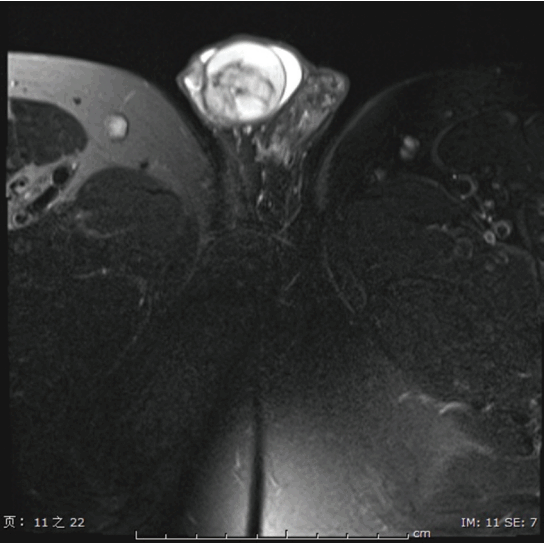
A 33-year-old male, visited the outpatient department because of a right testicular mass, detected by ultrasound during a routine health examination. Subsequently, the computed tomography scan was preformed, which showed a 3 cm×3 cm solid-cystic mass in the right testicle (Fig. 1). The patient then underwent unilateral orchidectomy, with the specimen analyzed by histological examination. No further adjuvant treatment was performed after diagnosis. The patient is currently disease-free 12 months after diagnosis. The patient was diagnosed as a rare case of MCST of the testicles, based on the histopathology, immunohistochemistry, and genetic analysis.
The surgical samples were fixed in 4% buffered formalin and embedded in paraffin. Sections were cut and stained with hematoxylin and eosin and reviewed by the authors. For immunohistochemical staining, 3-4-μm-thick sections were cut, and deparaffinized. Heat-induced antigen retrieval was performed with slides placing in 0.01 M of citrate buffer and 3% hydrogen peroxide methanol solution treatment was conducted for 30 minutes to eliminate nonspecific protein reactions. The antibodies are given as follows: vimentin (clone V9, 1:50), CD10 (clone 56C6, prediluted), β-catenin (clone 14, prediluted), cyclin D1 (clone SP4, 1:100), inhibin (clone R1, 1:20), calretinin (clone 5A5, 1:100), pan-cytokeratin (clone AE1/AE3, 1:100), cytokeratin (CK; clone Cam5.2, 1:200), hector battifora mesothelial epitope-1 (HBME-1; clone HBME-1, 1:100), WT-1 (clone 6F-H2, prediluted), sal-like protein 4 (SALL4; clone 6E3, 1:100), placental-like alkaline phosphatase (PLAP; clone 8B6, prediluted), octamer-binding protein 3/4 (OCT3/4; clone N1NK, 1:100), α-fetoprotetin (AFP; clone C3, 1:750), estrogen receptor (ER; clone 1D5, 1:50), progesterone receptor (PR; clone PgR636, 1:100), synaptophysin (Syn; clone SY38, 1:50), chromogranin A (CgA; clone LK2H10, 1:100), CD56 (clone 123C3.D5, 1:100), CD31 (clone JC70A, prediluted), CD34 (clone QBEnd/10, 1:250), erythroblast transformation specific related gene (ERG; clone EPR3864, 1:100), desmin (clone D33, 1:200), smooth muscle actin (SMA; clone 1A4, 1:400), epithelial membrane antigen (EMA; clone E29, 1:200), CD99 (clone 12E7, 1:20), and Ki-67 (clone MIB-1, 1:100). Binding of primary antibodies was visualized with an Envision two-step method. Positive and negative controls were included in this study. For genomic analysis, DNA was first extracted from the formalin-fixed, paraffin-embedded tumor blocks. Then polymerase chain reactions (PCRs) were performed to amplify exon 3 of catenin (cadherin-associated protein), beta 1 (CTNNB1) and exon 1 of forkhead box protein L2 (FOXL2) using following primer pairs: 5′-GATTTGATGGAGTTGGACATGG-3′ (CTNNB1 forward) and 5′-GCTACTTGTTCTTGAGTGAA-GG-3′ (CTNNB1 reverse); 5′-CCGCCACAACCTCAGCCTC-3′ (FOXL2 forward) and 5′-CGCCGGTAGTTGCCCTTCTC-3′ (FOXL2 reverse). Bidirectional Sanger sequencing of all PCR products was performed subsequently on a 3500 Genetic Analyzer (Life Technologies, Carlsbad, CA) using the BigDye Terminator v1.1 Cycle Sequencing Kit (Life Technologies), according to standard protocols.
In terms of gross anatomy, the mass was a well-circumscribed nodule measuring 3 cm in the largest diameter, with the cut surface being grey-white and solid. No typical macrocyst was observed. histopathologically, the tumor is composed of solid sheets of tumor cells and intervening bundles of hyalinized fibrous bands separating tumor clusters at low power view (Fig. 2A and B). In some regions, variable microcysts could be observed. At the high-power view, the uniform tumor cells contained granular eosinophilic cytoplasm with intracytoplasmic vacuoles, and round to oval nuclei with open chromatin and small nucleoli. There were no nuclear grooves and the nuclear membrane was irregular. There were no nuclear atypia or necrosis, with a low mitotic index. Immunohistochemically, the tumor cells had diffuse and strong cytoplasmic immunoreactivity for vimentin (Fig. 2C) and CD10 (Fig. 2D), as well as nuclear stains for β-catenin (Fig. 2E) and cyclin D1 (Fig. 2F), but no positive expression with sex cord markers (α-inhibin and calretinin), germ cell tumor markers (AFP, PLAP, Oct 3/4; and SALL4), and mesothelial markers (AE1/AE3, Cam 5.2, vimentin, WT-1, HBME-1, and calretinin). Other negative markers included hormone receptors (ER and PR), neuroendocrine markers (Syn, CgA, and CD56), vascular markers (CD31, CD34, and ERG), and other markers including desmin, SMA, EMA, CK7, and CD99. The Ki-67‒labeling index was about 10%. The result of sequencing showed that a heterozygous missense mutation (c.110C>G) in exon 3 of CTNNB1 was identified in this case (Fig. 3), resulting in amino acid changes. No mutation of FOXL2 was detected in the presented case (data not shown).
Discussion
Sex cord-stromal tumors occur in the gonads of both sexes, and they have only different frequency of occurrence of the individual subtypes [4]. According to the revised World Health Organization sex cord-stromal tumor classification, ovarian sex cord-stromal tumors were regrouped into the following clinicopathological entities: pure stromal tumors, pure sex cord tumors, and mixed sex cord-stromal tumors [5]. While testicular sex-stromal tumors include granulosa cell tumors, Leydig cell tumors, Sertoli cell tumors, and mixed and undifferentiated types [6]. The MCST is a recently identified and rare variant of the pure stromal tumor. In contrast to other sex-cord stromal tumors that are frequently identified in the gonads of both sexes, MCST appears to be restricted to the ovaries. In this report, we present the first case of MCST in the testicles, based on unique morphological features, immuno-profile, and molecular analysis.
MCST was first described in 2009 by Irving and Young [1]. Based on a series of 16 cases, Irving and Young [1] gave a detailed clinicopathological description of MCST as a new entity. The tumors showed distinctive histopathological appearance of solid, microcystic, or macrocystic patterns, as well as uniform or round cells with low mitotic index, which were separated by hyaline bands and fibrous plaques. These tumor cells were typically positive for vimentin, CD10, and β-catenin (nuclear expression), but negative for inhibin, calretinin, EMA, and other germ cell markers based in immunohistochemistry. Following the previous study, more cases with immunohistochemical and genetic analysis were presented [7,8].
Cyclin D1, known as one of the β-catenin regulated oncogenes, is often co-expressed with β-catenin in many malignancies. Cyclin D1 nuclear expression in MCST was reported recently, with the co-expression of cyclin D1 and β-catenin in the present case providing further evidence to support the involvement of the Wnt/β-catenin signaling pathway in the tumorognesis of MCST. In general, an immunohistochemistry panel recommended for the diagnosis of MCST is as follows: positivity for vimentin, CD10, β-catenin (nuclear expression), and cyclin D1 (nuclear expression); and negativity for inhibin, calretinin, and EMA. The immuno-profile for MCST diagnosis from all 31 previous cases are summarized in Table 1.
The β-catenin gene (CTNNB1) mutation, which causes the deregulation of β-TrCP–mediated β-catenin degradation and thus leads to the nuclear accumulation of β-catenin [2,9,10] mutation, was confirmed in most of the reported MCST cases in the literature based on sequencing and immunohistochemistry [11,12]. All the above findings suggest that the Wnt/β-catenin signaling pathway plays an important role in MCST tumorigenesis. In addition, among the 31 reported MCST cases, two were found to have clinic manifestations of familial adenomatous polyposis, as well as demonstrated allergic APC gene mutations [8,13], which led to a reduced ubiquitin-mediated destruction of β-catenin. These findings further emphasize the important role of the Wnt/β-catenin pathway in the development of MCST. Furthermore, MCST could be as a rare phenotype of familial adenomatous polyposis. In ovarian MCST, c.97T>C and c.101G>A are the most common loci of the CTNNB1 exon 3 mutation. In this study, the reported patient exhibited a rare point mutation of c.110C>G, which was rarely reported. More cases of MCST in the testicles should be collected to determine if c.110C>G of the CTNNB1 exon 3 is a preferential molecular abnormality in the testicle, which would contrast with the ovarian MCST.
MCST was once classified as “tumors of uncertain origin,” considering the pluripotency of the tumor cells, and then reclassified later as a new entity within the category of sex cord-stromal neoplasms. MCST demonstrates FOXL2 expression, which was thought to be highly sensitive and specific for sex cord-stromal tumors. However, a FOXL2 mutation was not detected in all MCST cases of the ovary [14]. In the present case, we did not identify a FOXL2 mutation.
The differential diagnosis of MCST in the testicles is similar to those occurring in the ovary, which include granulosa cell tumor, Sertoli-Leydig tumor, Steroid cell tumor, fibroma-thecoma, yolk sac tumor, and adenomatoid tumor. Adult granulosa cell tumor has oval cells, with nuclear grooves and Call-Exner bodies, shows immuno-activity for α-inhibin and calretinin, and harbors the FOXL2 gene mutation. Sertoli-Leydig tumor features round, oval, elongated, or irregular tubules in a fibrous stroma mixed with clusters of Leydig cells, with positivity for α-inhibin and calretinin. Steroid cell tumor is composed of cells, with abundant foamy pale cytoplasm and small nuclei. Cell clusters are separated by fibrous bands, and these tumors are immuno-active for α-inhibin and calretinin. Fibroma-thecoma shows estrogenic manifestations and immuno-activity for α-inhibin, calretinin, and CD56. Yolk sac tumor has a large mass size, elevated serum AFP, Schiller-Duval bodies, and immuno-activity for SALL4 and AFP. Besides the tumors mentioned above, mesothelial tumor was a common tumor that occurs in the testicle, which should be considered as a differential diagnosis. Adenomatoid tumor in the testicular area is a benign mesothelial tumor, which shows characteristic variably-sized tubular structures, with a pseudo-infiltrative growth pattern lined by bland, low-cuboidal epithelial cells, and immuno-activity for vimentin, calretinin, WT-1, HBME-1, and CK5/6 [15].
In summary, to the best of our knowledge, we presented the first case of a MCST in the testicle. MCST is a distinct clinical and pathologic entity within the category of sex cordstromal tumors, with the awareness of this rare entity being especially important for pathologists and surgeons.