Introduction
The 2022, World Health Organization (WHO) classification of adenocarcinoma of the urinary tract, including the urethra, listed uncommon Müllerian-derived carcinomas along with conventional adenocarcinomas not otherwise specified and enteric and mucinous types. Müllerian-type tumors are composed of clear cells and endometrioid adenocarcinomas originating from benign Müllerian precursors, such as endometriosis or Müllerianosis [1].
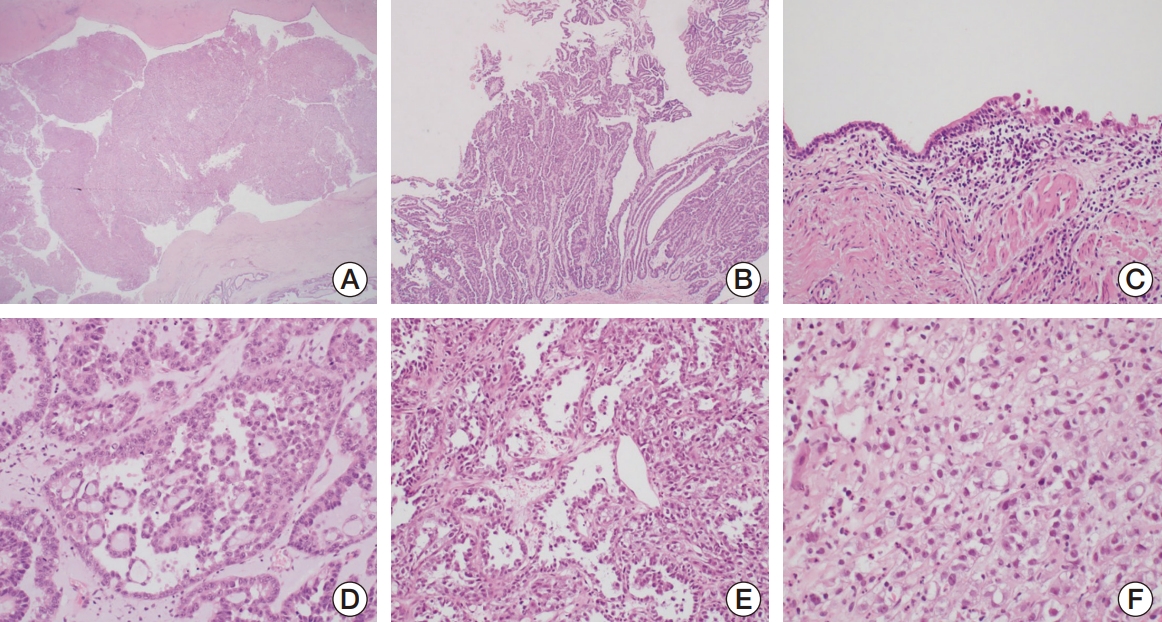
Clear cell adenocarcinoma (CCA) of the urinary tract is a rare malignancy, occurring predominantly in women. The female-to-male sex predilection of this tumor has been reported variably, ranging from 17:1 to 4:1 of in small series, with very few male cases [2]. CCA frequently involves the lower urinary tract in women, most commonly occurring in the urethra, where it may arise in the paraurethral ducts or diverticula [3]. This neoplasm is morphologically identical to the Müllerian-derived clear cell carcinoma of the female genital tract characterized by tubulocystic or papillary architecture with hobnail-like cells showing abundant clear or eosinophilic cytoplasm, moderate-to-marked nuclear pleomorphism, and frequent mitosis [1].
Immunohistologically tumor cells show positivity for cytokerain 7 (CK7), paired box 8 (PAX8), alpha-methylacyl-CoA racemase (AMACR), and p53 and negativity for GATA binding protein 3 (GATA3), p63, estrogen receptor (ER), and progesterone receptor (PR) [3]. The histogenesis of CCA in the urinary tract remains controversial, although several theories have been proposed. Currently, the most widely accepted theory is that this tumor is derived from the Müllerian remnant of the genitourinary system, as recognized by the 2022 WHO classification [1]. However, other theories insisting on a mesonephric remnant or urothelial origin have been reported, with supporting molecular and histological evidence [4,5].
Despite its aggressive behavior and poor prognosis, the molecular signature of urinary CCA and the strategies for improved clinical management have not been definitively established, owing to the paucity of cases. Previous studies have reported mutations in ATM, SMAD4, PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), KRAS, ARID1A (AT-rich interaction domain 1A), SMARCA4 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4) and the PI3K (phosphoinositide 3-kinase)/AKT/mTOR (mammalian target of rapamycin) pathway and chromatin modeling pathway have been proposed as pathogenic pathways [6-8]. In this study, we analyzed five cases of CCA of the urinary tract, especially the urethra. We performed whole exome sequencing (WES) and subsequent bioinformatics analyses to determine the mutational spectrum and possible pathogenic pathways of this rare neoplasm.
Materials and Methods
1. Patient selection and clinicopathologic review
We collected the data of a total of five patients who were diagnosed with CCA of the urinary tract at Seoul National University Hospital between January 1999 and December 2016. All cases were independently reviewed by two pathologists (K.C.M. and B.S.), including a genitourinary pathologist, who confirmed the diagnosis. If there was a disagreement on the microscopic evaluation, another pathologist was invited to review the slides for diagnostic consensus. Clinical data, including patient age, sex, and follow-up findings, were obtained from electronic medical records. This study was approved by the regional Institutional Review Board (No. H-2207-201-1346).
2. Immunohistochemistry
To confirm the diagnosis, representative slides of all cases were stained for CK7, PAX8, AMACR, napsin A, and HNF1 homeobox B (HNF1β). Only tumors with > 50% nuclear positivity for PAX8 were included. Additional staining for GATA3, p53, Ki-67, ARID1A, AMT, Smad4, β-catenin, phosphomTOR, phospho-p44/42 mitogen-activated protein kinase (MAPK), CHD4, PBRM1, programmed death-ligand 1 (PD-L1) (SP142 and 22C3) was also performed. Immunohistochemistry (IHC) was performed using an avidin-biotin-peroxidase detection system on a BenchMark ULTRA Stainer (Ventana, Tucson, AZ) according to the manufacturer’s recommendations. Used primary antibodies are described in S1 Table.
3. WES and bioinformatic analysis
Normal and cancer tissue were obtained from formalinfixed, paraffin embedded tissue blocks. DNA was extracted, and the quality of DNA was checked. Whole exome sequencing (WES) was conducted on a HiSeq 2500 platform (Illumina, San Diego, CA). Sequencing libraries were prepared, and adapter ligated DNA was amplified. Sequence was mapped to NCBI b37 human reference genome sequence, and BAM files were realigned. The MuTect2 algorithm was used to identify somatic variants which is allele depth of ≥ 10× and variant allele frequency of ≥ 10%, including single nucleotide variants (SNVs), small insertions and deletions (Indels).
Further filtering of pathogenicity was assessed using publicly available resources, cBioPortal, Catalogue Of Somatic Mutations In Cancer (COSMIC) and commercially available cancer panels (S2 Table) [9,10].
The variants absent in public database were excluded. The alterations of cancer-related genes were identified.
The final list of selected SNV and Indels in the present study was compared with additional cancer entities, such as endometrial carcinoma, serous carcinoma of the ovary, urothelial carcinoma of the bladder and clear cell renal cell carcinoma, using publicly available datasets from The Cancer Genome Atlas (TCGA). SNVs and clinicopathological data were retrieved from the cBioPortal platform [10] for comparison with the genetic alterations identified in this study.
Pathogenic pathways were analyzed using the method described above. Gene ontology enrichment analysis was used to identify biological processes, molecular functions, and cellular components [11]. Protein-protein interaction networks was assessed using STRING (http://string-db.org) to determine the putative pathogenic pathways.
Results
1. Clinical and histopathologic features of urethral CCA
The demographics, clinicopathologic characteristics, treatments, and outcomes of the five patients with CCA of the urinary tract are shown in Table 1. We found five CCA cases located in the urethra, out of 58 primary urethral carcinomas and 3,651 primary carcinomas of urinary tract between January 1999 and December 2016. All patients were female, and the median age at diagnosis was 62 years (range, 53 to 74 years). All tumors were located in the urethra, and in three cases, tumors developed in the proximal part. All patients underwent surgical interventions (radical cystectomy with vaginectomy, pelvic extension, and transurethral resection with pelvic lymph node dissection), and three of five patients received adjuvant chemotherapy or chemoradiation therapy. In this study, aggressive behavior of CCAs was observed, with rapid progression and poor prognosis. The average duration of follow-up was 23 months (range, 9 to 37 months), and all five patients experienced disease progression, namely exhibiting lung metastases (4/5), neck and mediastinal lymph node metastasis (1/5), peritoneal seeding (1/5), and local recurrence (1/5). Two patients died, and one died within year of diagnosis (11 months). Two patients had a history of breast cancer.
Histopathologic findings of CCA of the urinary tract are illustrated in Fig. 1. The tumors had clear and hobnail cells in a tubulocystic pattern, characteristic of CCA. Tumor architecture and nuclear pleomorphism varied among the cases. Nuclear pleomorphism was prominent in tumors with papillary and solid architectures. In four patients, an accompanying urethral diverticulum was observed. In three patients, the tumors had invaded the adjacent vagina, necessitating radical surgical treatment. Lymph node metastasis was present in three of the five patients with extensive involvement. This indicates that the cancer spreads to the lymph nodes and may have a higher risk of distant metastasis. All patients progressed to distant metastases, mostly to the lungs (4/5) or mediastinal lymph nodes (1/5).
2. IHC findings
The IHC staining results for several markers in the five patients are detailed in Table 2 and Fig. 2. All tumors exhibited strong nuclear positivity for PAX8, markers specific for tumors of the mesonephric (Wolffian) or Müllerian duct origin, and negativity for GATA3, suggesting non-urothelial origin. All tumors were positive for AMACR with variable cytoplasmic expression from focal weak positivity to strong positivity. In terms of markers of clear cell adenocarcinoma, all cases were positive for HNF1β (4/5) or Napsin A (2/5), with higher sensitivity for HNF1β. Ki-67, a marker of cell proliferation, showed variable expression ranging from 1%-70%.
3. Genetic alterations of cancer genes in urethral CCA
This study identified several genetic alterations that may play roles in the development of CCA (Table 3). Twenty-one SNVs and small Indels (< 50 bp) were identified, including 16 missense variants, one frameshift variant, two nonsense variants, one in-frame deletion, and one splice site variant. No identical recurrent mutations were detected among the variants. The set of identified cancer genes included seven driver mutations, including notable genetic alterations such as the oncogene KRAS p.Gly12Val (c.35G>T) variant, tumor suppressor gene PIK3R1 p.Arg557Gln (c.1670G>A), and ARID1A p.Trp1545*(c.4634G>A) variant.
4. Comparison with TCGA data of cancers of other origin
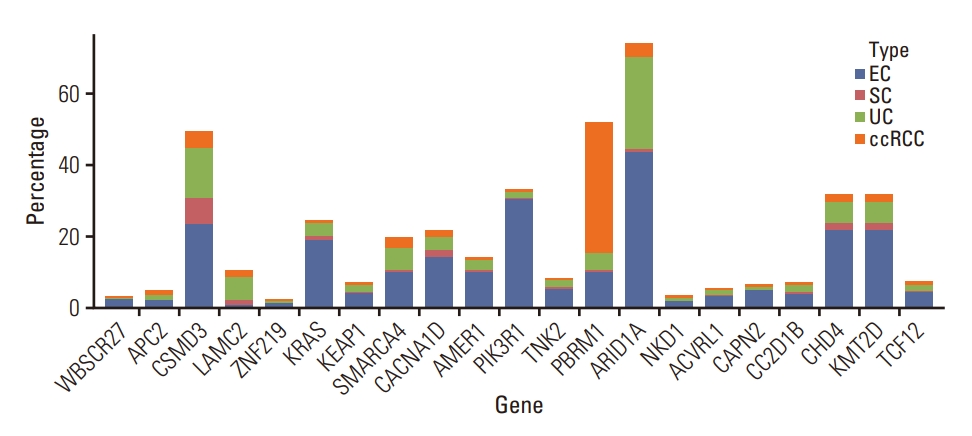
We compared the SNV alterations identified in our series of urethral CCAs with cancers of other origins, including endometrial adenocarcinoma of the uterus (n=517), serous adenocarcinoma of the ovary (n=525), urothelial carcinoma of the bladder (n=410), and clear cell renal cell carcinoma (n=402) from the TCGA platform. Fig. 3 illustrates the SNVs that showed the highest similarity between CCA and endometrial cancer of the uterus, with the highest overall frequency of the analyzed genes. ARID1A, PIK3R1, CSMD3, KRAS, CHD4, and KMT2D showed specific similarities between CCA and uterine endometrial adenocarcinoma.
5. Putative pathogenic pathways
1) Pathways, SNVs, and Indels
The pathogenic pathways of urethral CCA according to the identified 21 cancer gene alterations are shown in Table 4. The most notable molecular pathway associated with CCA was the chromatin remodeling pathway, activated by ARID1A, SMARCA4, ZNF219 (zinc finger protein 219), KMT2D (lysine methyltransferase 2D), and PBRM1 (polybromo 1) observed in three patients (patients 3-5). Another important molecular pathway was the PI3K/AKT/mTOR pathway activated by LAMC2 (laminin subunit gamma 2), KRAS, PIK3R1 (phosphoinositide-3-kinase regulatory subunit 1), and ACVRL1 (activin A receptor like type 1) gene alterations, observed in four patients (patients 2-5). In addition, the Keap1-Nrf2 pathway, which is activated by KEAP1 (kelch like ECH associated protein 1) tumor suppressor gene, and the MAPK signaling pathway, which is activated by mutations in the KRAS, CACNA1D (calcium voltage-gated channel subunit alpha1 D), ACVRL1, and TCF12 (transcription factor 12) gene, have been found to be associated with CCA. Other molecular pathways that have been implicated in the development of CCA include the Wnt/β-catenin pathway (APC2 [APC regulator of WNT signaling pathway 2], AMER1 [APC membrane recruitment protein 1], and NKD1 [NKD inhibitor of WNT signaling pathway 1]) and integrin-mediated cell adhesion pathway (CAPN2 [calpain 2]).
2) Gene ontology analysis
Gene ontology analysis revealed that some molecular functions and biological processes were enriched by altered genes in urethral CCA (S3-S5 Tables). Altered genes were associated with ATP-dependent chromatin remodeling activity (ARID1A, CHD4, and SMARCA4) and protein domain-specific binding (CACNA1D, KEAP1, KRAS, NKD1, PIK3R1, TCF12, and TNK2 [tyrosine kinase non receptor 2]) in molecular function and regulation of nucleotide-excision repair (ARID1A, PBRM1, and SMARCA4), positive regulation of cell differentiation (ACVRL1, ARID1A, KRAS, PBRM1, PIK3R1, SMARCA4, TCF12, and ZNF219), regulation of G0 to G1 transition (ARID1A, PBRM1, and SMARCA4), positive regulation of nucleic acid-templated transcription (ACVRL1, ARID1A, CHD4 [chromodomain helicase DNA binding protein 4], KMT2D, PBRM1, PIK3R1, SMARCA4, TCF-12, and ZNF219), and chromatin remodeling (ARID1A, CHD4, KMT2D, PBRM1, and SMARCA4) in biological processes.
3) Protein-protein interaction and network analysis
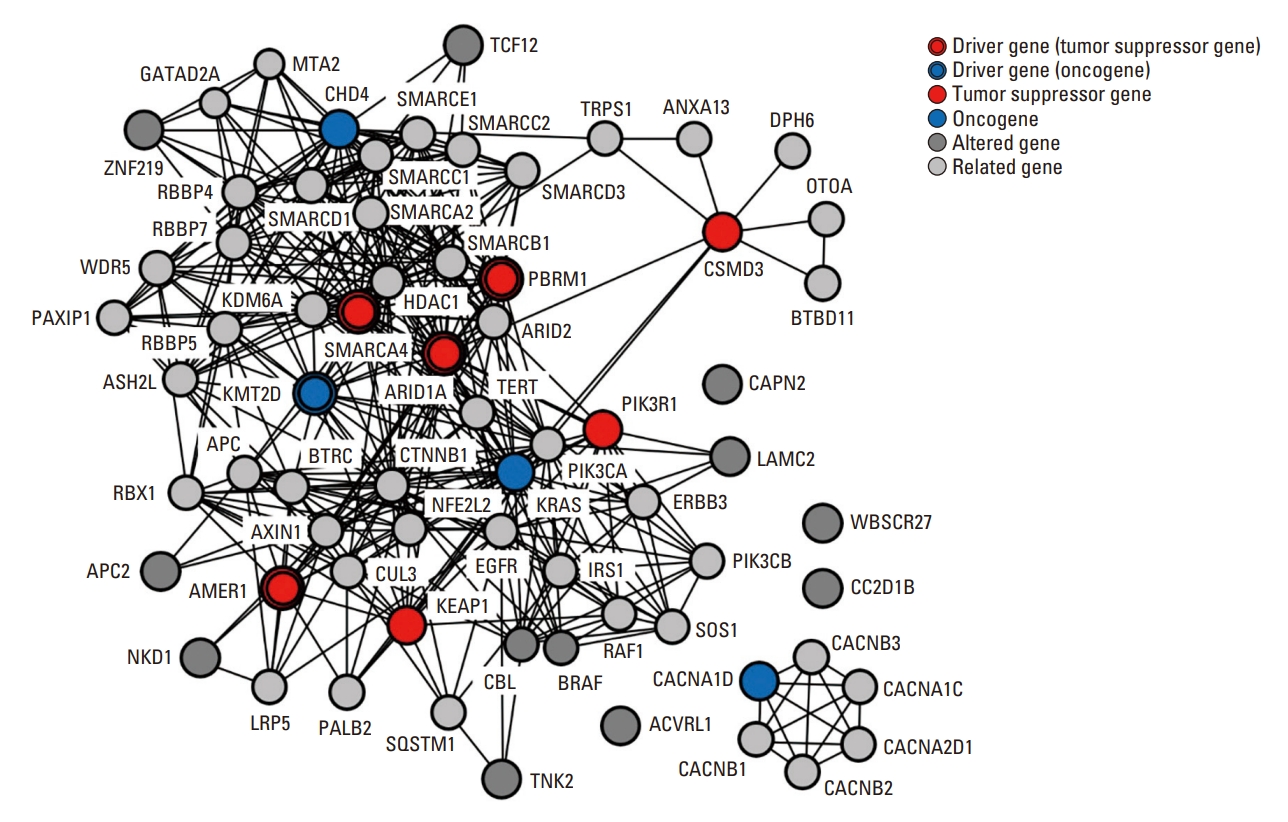
We conducted protein-protein interaction and network analyses based on the altered genes and their related genes (Fig. 4). To expand and assign weights to altered tumor suppressor genes (TSGs) or OGs, we included five related genes for each altered TSG or OG. As mentioned previously, AMER1, ARID1A, CSMD3 (CUB and Sushi multiple domains 3), KEAP1, PBRM1, PIK3R1, and SMARCA4 were regarded as TSGs, CACNA1D, CHD4, KMT2D, and KRAS were regarded as OGs. We included 76 genes to evaluate protein-protein interactions and performed network analysis. We found that the chromatin remodeling, PI3K/Akt/mTOR, and Wnt/β-catenin pathways were main pathogenic pathways in CCA of urethra.
6. IHC validation of genetic alteration and biomarkers
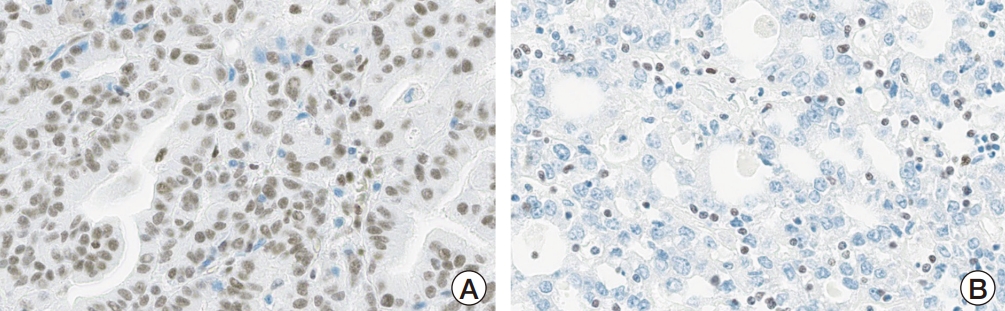
IHC staining results for validation of the protein expression of genetic alterations and putative pathways detected in our series, and previously reported mutations in urinary CCA are detailed in Table 5, Fig. 5, and S6 Fig. Loss of ARID1A expression was reported in three out of five cases, including patient 5 with ARID1A SNV alteration (p.Trp1545* [c.4634G>A] variant). Expression of CHD4 was identified in in two out of five cases, including patient 5 with CHD4 V1608I missense mutation (c.4822G>A, p.Val1608Ile). Loss of PBRM1 expression was not observed in PBRM1 mutant-patient 5 with strong nuclear positivity like overexpression, but patient 1 with wild type PBRM1 lacked protein expression in IHC. The expressions of phospho-mTOR were found in two patients (patients 3 and 5) with associated cancer-related gene alteration (KRAS and ACVRL1). Three patients (patients 3, 4 and 5) with KRAS, CACNA1D, ACVRL1, and TCF1 mutations reveals phospho-p44/42 MAPK positivity. Aberrant cytop-lasmic expressions (not nuclear expression; loss of membranous expression) of β-catenin were identified in two patients, including patient 4 with AMER1 mutation. Most patients (4/5) exhibited heterogenous p53 nuclear expression. Loss of ATM expression was identified in two, who also showed wild type p53 expression. The expression of SMAD4 associated with the transforming growth factor β was retained in most cases (4/5). PD-L1 SP142 and 22C3 were not expressed in any of the patients.
Discussion
Urinary tract CCAs are rare. Owing to the rarity of the entity, the nature and prognosis of this tumor remains unclear. The theory of Müllerian origin is generally accepted, and the tumor shows female predominance with an M:F ratio of 1:4-6 [1,2,12]. Pathologically, CCA presents as a polypoid, papillary, or ulcerative mass. The tumor cells showed moderate-to-severe atypia with variable mitosis and necrosis, and papillary, tubulocystic, and/or solid growth patterns. The immunophenotype of urinary CCA varies among patients. Tumor cells are usually positive for PAX-2, PAX-8, epithelial membrane antigen, CK (AE1/AE3), CAM 5.2, CK7, AMACR, and p53, and negative for GATA3, p63, ER, and PR, CK20, carbohydrate antigen 125 and variable levels of Ki-67 index [3]. In the majority of urinary CCA cases, aggressive behaviors have been reported such as high local recurrence, lymph node metastasis, distant metastasis, and disease-related mortality, with a 5-year survival rate of 40% [13]. Treatment includes surgical resection with or without adjuvant chemotherapy or radiation therapy. Brachytherapy has been utilized as well [13]. No targeted therapies have been introduced because the pathogenic alterations or actionable targets have not yet been well established. Immunotherapy with immune checkpoint inhibitors is not considered because of the lack of known data on PD-L1 expression. The molecular signature of urinary CCA has not been definitively established, owing to the paucity of cases. A previous genetic study of one case reported truncating mutations in the ATM, copy number loss at the SMAD4 and ARID2 loci, and gene fusion candidates, including ANKRD28-FNDC3B [8]. In another series of four cases, recurrent pathogenic PIK3CA (p.E545K) and KRAS (p.G12D) variants (3/4), an APC (p.S2310X) variant (1/4), a TP53 (p.R273C) variant (1/4), and MYC amplification (1/4) were identified; the authors suggested that the activation of the PI3K/AKT/mTOR pathway was a possible underlying oncogenic events [7]. In recent studies, mutations in KRAS and chromatin modeling genes, such as ARID1A, ARD1B, and SMARCA4, were reported to be common finding of female patients with genital tract CCA [6].
In this study, we examined the data of five patients with urinary CCA, all of which occurred in the urethra, not only in terms of the clinical and pathological features but also in terms of the molecular biological features to explore the nature of this rare entity. In our case series of single center for 18 years, CCA was a rare histologic type, accounting for 8% of primary urethral carcinomas and 0.1% of all urinary tract malignancies. CCA showed an advanced stage from the time of diagnosis and aggressive behavior with rapid clinical progression despite the combination of surgical resection and brachytherapy in most cases, indicating the need for careful monitoring of patients with urethral CCA as well as the increased use of aggressive treatment measures such as chemotherapy and chemoradiation.
IHC for p53, ATM, and SMAD4 was performed to assess the protein expression of previously reported mutations in urinary CCA [6-8]. Our series of CCA showed no SNV or INDEL variants of these mutations on WES, but some patients showed mutant-type expressions on IHC. These findings suggest the possibility of genetic alterations affecting protein expression, such as CNVs, in addition to the point mutations, SNVs or Indels, that we examined. ATM and TP53 function together in the DNA damage response pathway, and there is a negative correlation between ATM and TP53 mutations in lung adenocarcinoma [14]. Loss of ATM expression with the wild-type p53 pattern was observed in two patients in our series. PD-L1 SP142 and 22C3 expressions, which should be considered before starting immunotherapy, were negative in all cases.
WES and bioinformatics analyses were performed to determine the genetic and possible pathogenic alterations. On comparing the identified mutation spectrum of urethral CCA to the TCGA data of endometrial, ovarian, bladder, and kidney tumors, which are differential diagnoses owing to similarities in location and histologic findings, CCA showed the highest similarity to endometrial adenocarcinoma. Genetic variants reported in previous studies, such as PIKC3CA and ARID1A [6,7,15], show a particularly high frequency in endometrial cancer, and driver genes such as CSMD3, CHD4, and KMT2D also show significant similarity to endometrial cancer. Although CSMD3 and ARID1A are also frequently altered in urothelial carcinoma or bladder cancer, the absence of a TERT promoter mutation, the most prevalent mutation in urothelial carcinoma [16], indicates the non-urothelial origin of urethral CCA. Urinary CCAs are known to arise from pre-existing Müllerian precursors within the urinary tract [1], and our molecular data support the theory of Müllerian origin.
We found several TSGs and OGs, some of which have been identified in previous studies (KRAS, ARID1A, SMARCA4) [6,7,15]. In patient 3, we identified KRAS G12V missense mutation (c.35G>T, p.Gly12Val), which is a well-known oncogene in the colorectal, pancreatic, lung, ovarian, and biliary tract cancers [9,17]. KRAS is a small GTPase that regulates many cellular responses through the MAPK, PI3K/AKT/mTOR, and RAS signaling pathways. Oncogenic activation of KRAS results in cell proliferation or immortality, angiogenesis, invasion, metastasis, changes in cellular energetics, and escape from apoptosis [18]. In patient 4, two TSGs, AMER1 and PIK3R1 were observed. The AMER1 encodes APC membrane recruitment protein 1 and is involved in β-catenin, thus inhibiting the Wnt/β-catenin pathway [19]. An AMER1 E752K missense variant (c.2254G>A, p.Glu752Lys) was also identified. AMER1 competes with NRF2 for binding to KEAP1. Interestingly, patient 4 harbored a KEAP1 mutation (c.1085G>A, p.Arg362Gln). KEAP1 is regarded as a TSG and is thought to suppress the expression of survival genes by inhibiting NRF2 [20]. ML385, an NRF2 inhibiting molecule, shows specificity and selectivity for NRF2-addicted non–small cell lung cancer cells with KEAP1 mutations and significant antitumor activity in combination with carboplatin [21]. There was a possibility of NRF2 activation in patient 4, and targeted therapy for NRF2 could have had a therapeutic effect. PIK3R1 is a component of the PI3K/AKT/mTOR pathway, which regulates cell proliferation, apoptosis, and metabolism [22]. In the present case, there was PIK3R1 R557Q missense mutation (c.1670G>A, p.Arg557Gln). As the inactivation of PIK3R1 could lead to mTOR pathway activity, drugs targeting the PI3K/AKT/mTOR pathway, such as everolimus and temsirolimus, would be beneficial. In patient 5, we identified PBRM1 and ARID1A mutations associated with chromatin remodeling, and CHD4 and KMT2D mutations associated with histone remodeling. A PBRM1 frameshift deletion (c.3245delG, p.Gly1082fs) was also identified. PBRM1 is a subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex and is involved in chromatin remodeling, thus affecting the expression of various genes [23]. PBRM1 mutations lead to genomic instability and mutations, impaired tumor growth suppression, tumor-promoting inflammation, and escape from the immune response to cancer [23]. In our study, the loss of PBRM1 expression was not validated in patient 5 with strong nuclear positivity. Gad et al. [24] recently reported increased PBRM1 expression with uniform and strong nuclear staining in Von Hippel-Lindau disease-associated clear cell renal cell carcinoma and a PBRM1 mutant case showing retained protein expression. Additionally, an ARID1A W1545* nonsense mutation (c.4634G>A, p.Trp1545*) was observed. ARID1A is the most commonly altered gene reported in prior genetic studies on urinary CCA [6,7,15]. They are also found in endometrial adenocarcinomas, lung adenocarcinomas, colon cancer, bladder urothelial carcinoma, and breast cancer [9]. In this study, the loss of ARID1A expression was validated in patient 5 as well as other two patients by IHC. ARID1A-deficient CCA in our series (patients 1, 2 and 5) showed no distinguishable clinical or pathological characteristics in common. ARID1A is a member of the SWI/SNF and chromatin remodeling complex and acts as a tumor suppressor gene [23]. ARID1A mutation is associated with genome instability and mutations, growth suppression, metastasis, and escaping. The inactivation of the SWI/SNF complex is considered a potential biomarker for various targeted drugs. Loss of ARID1A function sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitors and combined immunotherapy is recommended for ARID1A-deficient carcinomas. Ongoing studies involving ATR inhibitors have shown promising results, making ARID1A an attractive candidate for targeted therapy [25]. Given that a recent clinical trial testing ATR and PARP inhibitors in gynecologic cancers assessed ARID1A status by IHC followed by retrospective mutational analysis [26], and that ARID1A protein loss was observed in three out of five CCA cases in our study, the screening test by IHC for ARID1A in urinary CCA would be a cost-effective evaluation for targeted therapy. In addition, a CHD4 V1608I missense mutation (c.4822G>A, p.Val1608Ile) was identified. CHD4 is involved in histone deacetylation and dysfunction of this gene results in genomic instability and mutations. CHD4 exerts both oncogenic and tumor-suppressive effects and could be a therapeutic target [27]. In this study, the CHD4 expression was validated in patient 5 as well as another patient (patient 3) by IHC. In addition, a KMT2D S771F missense mutation (c.2312C>A, p.Ser771Tyr) was identified. KMT2D is a histone-lysine N-methyltransferase that targets H3 lysine 4 and affects the epigenetic regulation of various genes. KMT2D has both oncogenic and tumor-suppressive roles and is involved in invasion, metastasis, cell proliferation, immortality, and changes in cellular energetics [28].
Our data indicate that MAPK signaling pathway (KRAS, CACNA1D, ACVRL1, and TCF12), PI3K/AKT/mTOR pathway (KRAS, LAMC2, PIK3R1, and ACVRL1), Wnt/β-catenin pathway (APC2, AMER1, and NKD1), and chromatin remodeling (ARID1A, SMARCA4, ZNF219, KMT2D, and PBRM1) could be the pathogenic pathways related to CCA of the urinary tract, results of bioinformatics analyses supported these findings. The most notable molecular pathway of urinary CCA was the chromatin remodeling pathway with many significantly associated molecular functions and biological processes in gene ontology analysis, such as ATP-dependent chromatin remodeling activity (p=0.00000701), histone binding (p=0.00301) and others. Ortiz-Bruchle et al. [6] recently proposed the chromatin remodeling pathway as a pathognomonic pathway for urinary CCA in association with frequent ARID1A mutations in their study. We validated the activation of the suggested putative pathways by IHC, and substantial protein expression was identified in association with genetic mutations affecting the pathway (phospho-mTOR [2/5], phospho-p44/42 MAPK [3/5], β-catenin [2/5], ARID1A [3/5], CHD4 [2/5]). As shown in Fig. 5, various mutated genes identified in this study were grouped into chromatin remodeling, PI3K/AKT/mTOR pathway, Wnt/β-catenin pathway, supported by protein expression assessed by IHC. Our results suggest that therapies targeting these pathogenic pathways may be beneficial for patient management.
Discordance between IHC and mutational status was noted in some cases, such as wild type with altered protein expression (ARID1A in patients 1 and 2, CHD4 in patient 3, PBRM1 in patient 1) and mutant with not altered protein expression (PBRM1 in patient 5). Altered IHC without mutation could be explained by loss of heterozygosity, mutations in non-coding regions, and post-transcriptional and/or post-translational mechanisms [29]. Not altered protein expression in mutated cases could be explained by late truncating mutation where mRNA is not undergoing nonsense mediated decay [30]. Further evaluations were not performed due to methodological reason.
Interestingly, we also identified cancer-related genes associated with these pathogenic pathways. Genetic alterations in the MAPK signaling pathway included CACNA1D in patient 4 and ACVRL1, TCF12 in patient 5. CACNA1D mutations have also been found in patients with malignant melanoma, lung squamous cell carcinoma, and gastric cancer [9,17]. Genetic alterations in the PI3K/AKT/mTOR pathway were identified in patient 2 (LAMC2). LAMC2 mutations been reported in ovarian, lung, and pancreatic cancers. Genetic mutations involved in Wnt/β-catenin pathway was found in patients 2 (APC2) and 5 (NKD1). APC2 mutations have been identified in thyroid, malignant melanoma, and colorectal, bladder, and liver cancers. In addition, NKD1 mutations have been found in liver cancer and head and neck squamous cell carcinoma. Genetic alterations in chromatin remodeling were identified in patients 3 (ZNF219) and 4 (SMARCA4). ZNF219 mutations have been identified in patients with colorectal, thyroid, stomach, endometrial, and endometrial cancer [9,17]. SMARCA4 mutations have been reported in patients with lung, colorectal, endometrial, bladder, urothelial, and breast cancers. As shown in Fig. 5, various mutated genes identified in this study were grouped into chromatin remodeling, PI3K/AKT/mTOR pathway, Wnt/β-catenin pathway. Our results suggest that therapies targeting these pathogenic pathways may be beneficial for patient management.
This study had some limitations. First, although we collected data from a relatively large sample of CCA of the urinary tract, the number of cases was not sufficient for statistical analysis of clinicopathologic or pathognomonic correlations. In addition, in all our patients, the CCA arose in the urethra, so our findings cannot be applied to CCA arising from other parts of the urinary tract. Second, we performed WES and bioinformatics studies, and proposed several pathogenic pathways related to the pathogenesis of CCA based on genetic analyses. However, transcriptomic or proteomic studies other than IHC are needed to validate the oncogenic or tumor-suppressive roles of the driver genes. Third, we identified the actionable targets for CCA, but studies of cell lines, organoids, and animal models are needed to determine the clinical application of these targets.
In summary, we identified important driver gene mutations (AMER1, ARID1A, CHD4, KMT2D, KRAS, PBRM1, and PIK3R1) and pathogenic pathways (chromatin remodeling pathway, MAPK signaling pathway, PI3K/AKT/mTOR pathway and Wnt/β-catenin pathway) involved in CCA of the urethra. Owing to the rarity of the disease, further multicenter studies or meta-analyses are needed to validate our results. We hope that our results will contribute to enhance patient management.