Introduction
Ovarian cancer is the fifth leading cause of cancer deaths in women, and it is the most lethal of all gynecologic cancers [1]. In China, ovarian cancer is the third common female reproductive cancer [2]. Epithelial ovarian cancer (EOC) accounts for ~90% of all ovarian malignancies. The 5-year survival rate of EOC is ~45%, and it declines to 35% in the patients diagnosed at advanced stage due to lacking of specific symptoms and deficiency in screening programs [3,4]. Currently, surgery followed by platinum-based chemotherapy (e.g., cisplatin) is the standard treatment for EOC [5]. Despite all efforts to surgery and chemotherapy over last two decades, EOC is still associated with a high mortality and morbidity rate. The chief reason for the high mortality rate from ovarian cancer is chemoresistance. Therefore, it is critical to unravel the underlying mechanism contributing to chemoresistance in ovarian cancer, thus enhancing the sensitivity to platinum-based chemotherapy.
Long non-coding RNAs (lncRNAs) are a group of non-coding RNAs which are more than 200 nucleotides in length. Growing evidence has indicated that lncRNAs were implicated in a variety of biological processes by regulating gene expression at transcriptional and post-transcriptional levels [4,6]. Forinstance, the competitive endogenous RNA (ceRNA) hypothesis suggests that lncRNA acts as a competitive sponge of microRNA (miRNA) and inhibits miRNA function, thus modulating miRNA target gene expression. Recently, emerging evidence has demonstrated that lncRNAs are deregulated in tumor tissues and closely correlated with cancer initiation, progression and chemoresistance, suggesting that lncRNAs could act as diagnostic biomarkers and therapeutic targets in human cancers [4,7].
Colon cancer-associated transcript 1 (CCAT1) with 2,628 bp in length was located on chromosome 8q24.21. It was originally identified as an oncogenic lncRNA in colorectal cancer [8]. Following studies have revealed that CCAT1 is significantly elevated in a number of tumor types, such as gastric cancer, gallbladder cancer (GBC), breast cancer, lung cancer, hepatocellular cancer (HCC), and ovarian cancer [8-10]. Microarray analysis coupled with Quantitative reverse transcription polymerase chain reaction (qRT-PCR) showed that CCAT1 is up-regulated in high metastatic EOC cells, suggesting that CCAT1 is involved in ovarian cancer metastasis [11]. Moreover, previous researches have reported that CCAT1 plays a crucial role in modulating chemoresistance in cancer cells. For instance, CCAT1 sponges lec-7c to promote chemoresistance in docetaxel-resistant lung cancer cells [7]. It also regulates paclitaxel sensitivity in nasopharyngeal carcinoma cells via miR-181a/CPEB2 axis [12]. However, the mechanism by which CCAT1 modulates cisplatin resistance in EOC remains uninvestigated.
In this study, we studied the functional role of CCAT1 in cisplatin-resistant ovarian cancer cells. The results showed that CCAT1 was highly expressed in cisplatin-resistant A2780/DDP cells. CCAT1/miR-454/survivin axis implicated in mediating cisplatin resistance of ovarian cancer cells. Understanding the mechanism of chemoresistance may provide new insight into targeted therapeutic strategies of ovarian cancer.
Materials and Methods
1. Reagents and antibodies
Cisplatin was obtained from Sigma-Aldrich (St. Louis, MO). The shRNAs including both negative control and specific to CCAT1 were purchased from GenePharma (Jiangsu, China). Mimics control (miR-NC), miR-454 mimics, inhibitor control and miR-454 inhibitor were purchased from GenePharma. For survivin overexpression, the full length of survivin cDNA was cloned into pcDNA3.1 vector. Following antibodies were used in this study: anti-Bcl-2 (#2872), antiBax (#2774), anti-survivin (#2808), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; #2118) were purchased from Cell Signaling Technology (Beverly, MA). Lipofectamine 2000 transfection reagent was purchased from Thermo Fisher Scientific (Waltham, MA).
2. Cell culture and transfection
Human ovarian cancer cell lines A2780 and SKOV3 cells were obtained from Cell Bank of Type Culture Collection, Chinese Academy of Science (Shanghai, China). DDP-resistant A2780/DDP and SKOV3/DDP cells were generated as previously described [13]. In brief, original parental A2780 or SKOV3 cells were exposed to IC50 concentration of cisplatin. These cells were cultured in medium containing cisplatin for at least 6 months. Cells were cultured in RPMI-1640 medium supplemented with 10% FBS (Thermo Fisher Scientific), 100 μg penicillin and 100 U/mL streptomycin. Cultures were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C. For knockdown and overexpression experiments, A2780, A2780/DDP, SKOV3, or SKOV3/DDP cells were transfected with shRNA/mimics/inhibitor/plasmid using Lipofectamine 2000 transfection reagent according to the manufacturer’s instruction. Cells were harvested for qRTPCR analysis at 48 hours post-transfection.
3. MTT assay
Cell viability was monitored by MTT assay. A2780, A2780/DDP, SKOV3, and SKOV3/DDP cells were seeded in 96-well plates 24 hours prior to treatment. Cells were then treated with different doses (0, 5, 10, 20, 40, and 80 μM) of cisplatin for 24 hours. A total of 20 μL MTT was added into each well and incubated for 4 hours at 37°C. One hundred fifty microliters of dimethyl sulfoxide was then added into each well to resolve MTT formazan crystals. Absorbance was measured at a wavelength of 490 nm using Model 680 micro-plate reader (Bio-Rad Laboratories, Hercules, CA).
4. qRT-PCR
Total RNA was isolated fromA2780, A2780/DDP, SKOV3 and SKOV3/DDP cells using Trizol reagent (TaKaRa, Dalian, China) and reverse transcribed with Advantage RT-for-PCR Kit (TaKaRa). cDNA were used as templates for qRT-PCR with specific primers. The stem-loop RT primers (RiboBio Co., Guangzhou, China) were used to detect miR-454 or U6. Other primers used in this study were shown as following: CCAT1, sense, 5'-ATGGCTATGCAAGTCCCAAC-3', antisense, 5'-GCAGAAAGGCCAGTGCTTAC-3'; Bcl-2, sense, 5'-GATGACTGAGTACCTGAACC-3', antisense, 5'-AGTTCCACAAAGGCATCC-3'; Bax, sense, 5'-GCCCTTTTCTACTTTGCCAGC-3', antisense, 5'-CTGGAGACAGGGACATCAGT-3'; survivin, sense, 5'-GGACCACCGCATCTCTACAT-3', antisense, 5'-CTTTCTCCGCAGTTTCCTCA-3'. The mRNA level of the target gene was analyzed using Power SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific) according to the manufacturer’s instruction (n=3, each in triplicates). GAPDH or U6 was used as an internal control. The specificity of the fluorescent signal was verified by both gel electrophoresis and melting curve. The expression level of the target gene was determined using 2-ΔΔCT method.
5. Annexin-V-FITC/propidium iodide staining
Annexin-V-FITC/propidium iodide (PI) staining was performed using Anne-xin-V-FITC/PI apoptosis assay kit (BD Biosciences, San Diego, CA) according to the manufacturer’s instruction. In brief, cells were resuspended in binding buffer, and incubated with 5 μL Annexin-V-FITC reagent and 5 μL PI at room temperature for 15 minutes. The samples were analyzed by flow cytometry with BD FACSCalibur (BD Biosciences).
6. Western blotting
Protein lysates from A2780, A2780/DDP, SKOV3, and SKOV3/DDP cells and tumor tissues were prepared in RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktail (Pierce, Thermo Fisher Scientific). Total protein concentration was determined by Bradford Protein Assay Kit (Bio-Rad). Protein lysate were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto PVDF membrane (Bio-Rad) for western blotting analysis. The membrane was blocked with blocking reagent (5% non-fat milk), followed by overnight incubation with primary antibody at 4°C. The membrane was then incubated with respective horseradish peroxidase–conjugated secondary antibody. ECL western blotting detection reagents (Beyotime, Haimen, China) were used for protein detection.
7. Dual luciferase reporter assay
The online prediction database Starbase (http://starbase.sysu.edu.cn/index.php) and TargetScan (http://www.targetscan.org/vert_71/) were used to predict the potential miRNA target of CCAT1 and downstream targets of miR-454, respectively. The wild type (WT) and mutant (MUT) miR-454 binding sequence included fragments of CCAT1 or 3′-untranslated region (3′-UTR) of survivin were cloned into psiCHECK-2 vector (Promega, Madison, WI). The dual luciferase reporter assay was performed 48 hours post-transfection by the use of dual luciferase assay system (Promega) according to the manufacturer’s instructions. In brief, Cells were co-transfected with CCAT1-WT/MUT or survivin-WT/MUT and mimics con or miR-454 mimics using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific). The luciferase activity of each sample was normalized to the Renilla luciferase activity.
8. Xenograft study
Female BALB/c nude mice were purchased from the Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). All mice were housed in a controlled environment with 12 hours light/12 hours darkness cycles. A2780 or A2780/DDP cells were transfected with scramble shRNA (negative control [NC]) or sh-CCAT1. These cells were then implanted subcutaneously into the right flank of mice. At 7 days after inoculation, mice in A2780 and A2780/DDP groups received 5 mg/kg and 10 mg/kg cisplatin treatment every 3 days, respectively. Animals were sacrificed at 30 days following treatment and tumors were collected. The length (l) and width (w) of the tumors were measured by a digital caliper. The tumor volume (V) was calculated using the formula: V=lw2/2.
9. Statistical analysis
All experiments were performed at least three times. Data are expressed as the means±standard deviation. Statistical analysis was performed by one-way ANOVA followed by Tukey’s post-test. Student’s t test was used for paired comparisons in selected experiments. Statistical analysis was performed using the SPSS ver. 17.0 (SPSS Inc., Chicago, IL). p < 0.05 was considered statistically significant.
Results
1. Knockdown of CCAT1 enhances sensitivity to cisplatin in ovarian cancer cells
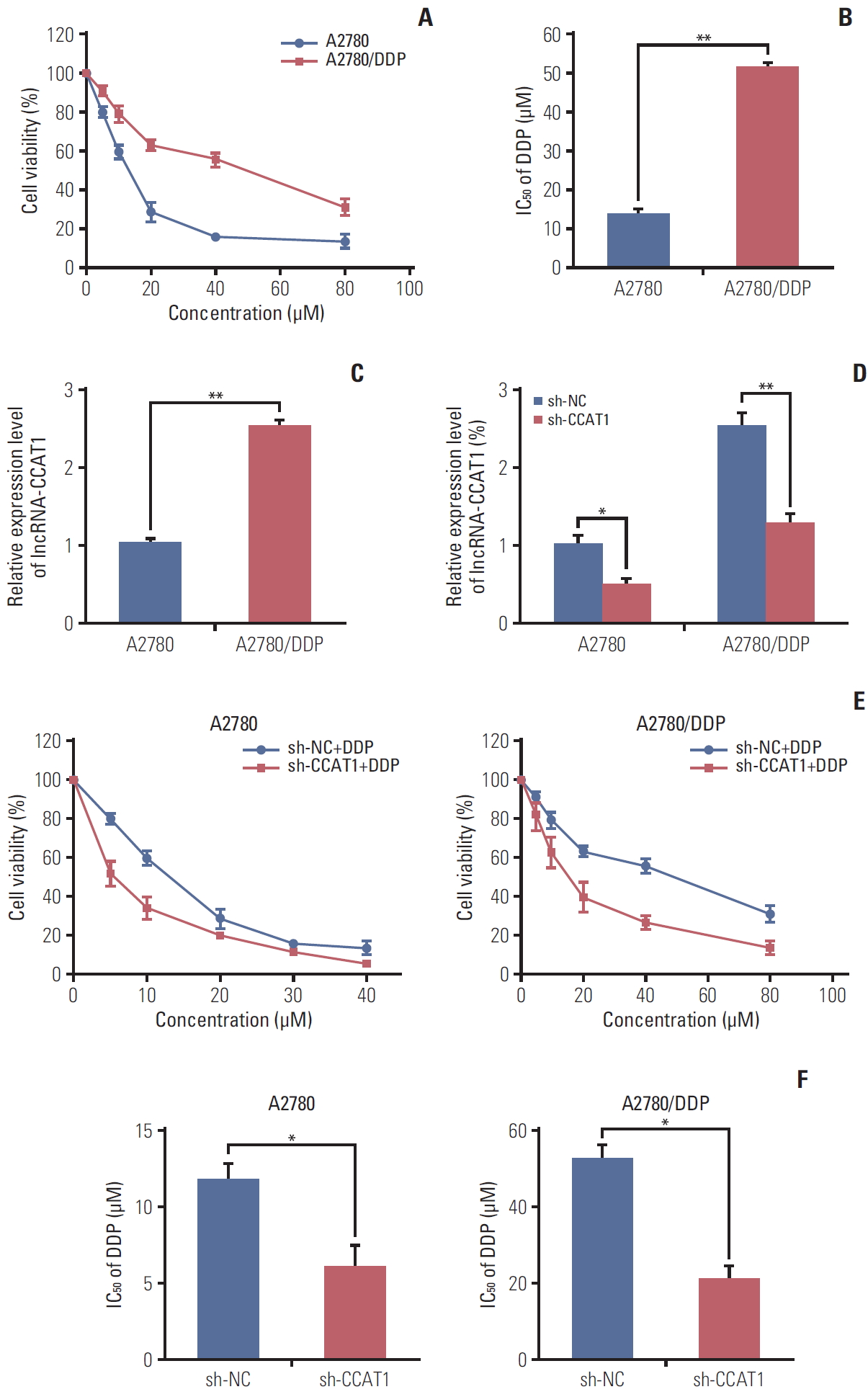
To explore the biological function of CCAT1 in ovarian cancer cells, cisplatin-resistant A2780 cell line, namely A2780/DDP cell, was generated. We compared the expression of CCAT1 in cisplatin-sensitive A2780 and cisplatin-resistant A2780/DDP cells. The cisplatin-resistance phenotype of A2780/DDP cells was firstly confirmed by MTT assay. Compared with the parental A2780 cells, A2780/DDP cell viability was significantly higher upon cisplatin treatment (Fig. 1A). The IC50 value of cisplatin in A2780 cells was 14.0 μM, and it increased to 51.6 μM in A2780/DDP cells, suggesting that A2780/DDP cells were more resistant to cisplatin (Fig. 1B). qRT-PCR was then carried out to determine the expression level of CCAT1 in these two cell lines. As presented in Fig. 1C, more than a 2.5-fold increase was found in CCAT1 level in A2780/DDP cells when compared with the parental cell line. Taken together, CCAT1 is up-regulated in cisplatin-resistant A2780/DDP cells, suggesting that CCAT1 might contribute to cisplatin-resistance in EOC.
We further investigated the effect of CCAT1 on cisplatin resistance in vitro. As shown in Fig. 1D, shRNA-mediated silencing of CCAT1 successfully decreased CCAT1 expression in both A2780 and A2780/DDP cells (Fig. 1D). MTT assays revealed that down-regulation of CCAT1 significantly enhanced cisplatin sensitivity in A2780 and A2780/DDP cells (Fig. 1E), presented by lower IC50 value (Fig. 1E and F). The IC50 of cisplatin in sh-CCAT1 transfected A2780 cells decreased by ~40% when compared with corresponding control. In A2780/DDP cells, knockdown of CCAT1 decreased IC50 by ~60% compared with control group (Fig. 1F). These data revealed that down-regulation of CCAT1 enhances A2780 and A2780/DDP cell susceptibility to cisplatin. Consistently, similar results were observed in SKOV3 and SKOV3/DDP cells. As shown in S1 Fig., CCAT1 was significantly elevated in cisplatin-resistant SKOV3/DDP cells compared to parental SKOV3 cells (S1A-S1C Fig.). Knockdown of CCAT1 enhanced the sensitivity to cisplatin in both SKOV3 and SKOV3/DDP cells, and the IC50 value of cisplatin was decreased in CCAT1 depleting cells compared to corresponding control cells (S1D-F Fig.). Collectively, these findings indicate that knockdown of CCAT1 enhances sensitivity to cisplatin in ovarian cancer cells.
2. Knockdown of CCAT1 modulates sensitivity of ovarian cancer cells to cisplatin via promoting cell apoptosis
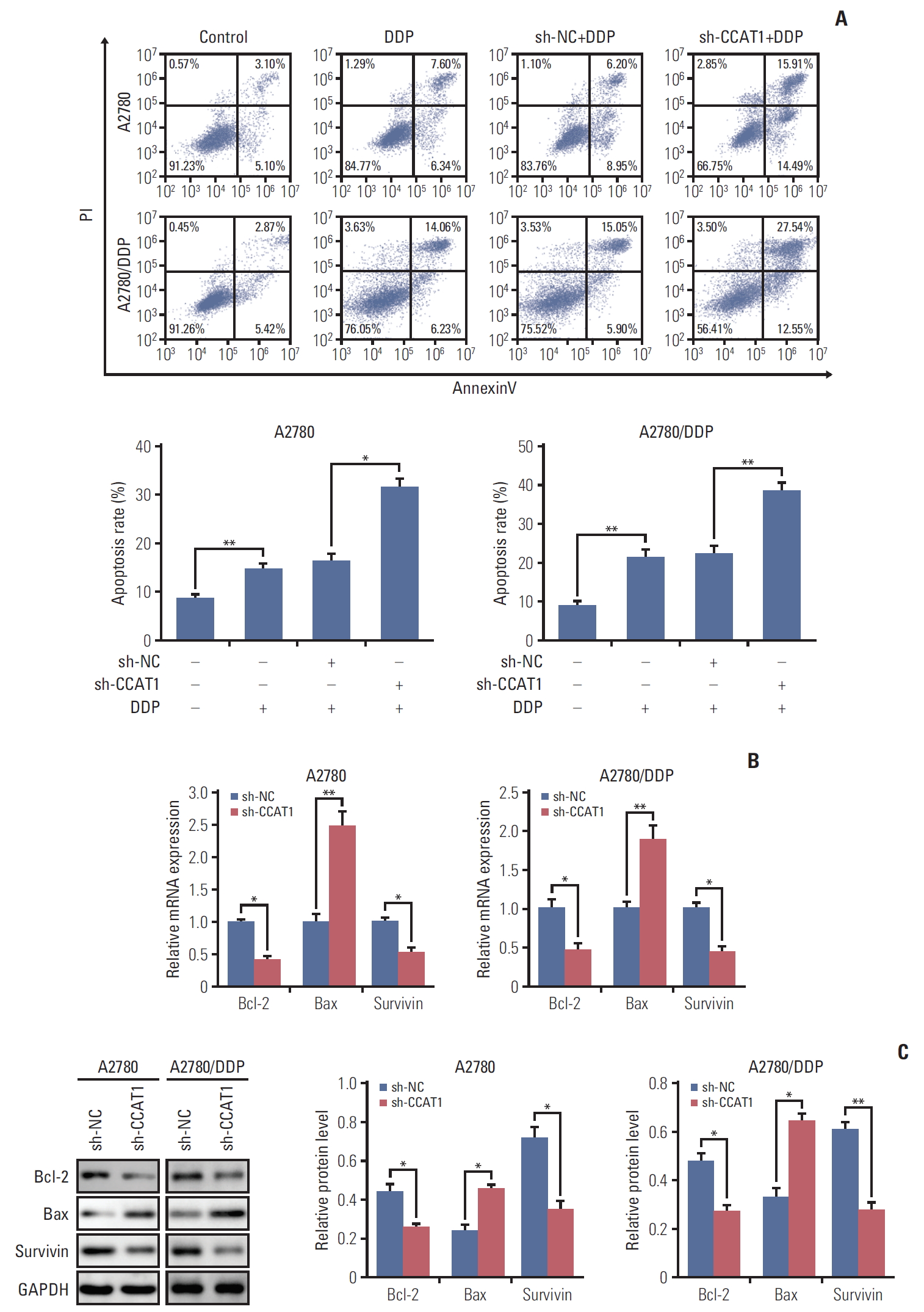
As mentioned above, knockdown of CCAT1 decreased the viability of A2780 and A2780/DDP cells upon cisplatin treatment. We thus test whether sh-CCAT1 induced growth inhibition via promoting apoptosis. A2780 and A2780/DDP cells were transfected with sh-CCAT1, and treated with cisplatin at 8 μM (A2780) or 20 μM (A2780/DDP) for 24 hours, followed by Annexin V-FITC/PI staining. As shown in Fig. 2A, silencing of CCAT1 significantly promoted cisplatininduced apoptosis in both A2780 and A2780/DDP cells. Expression of apoptosis-related genes was further detected by qRT-PCR and Western blotting. Silencing of CCAT1 down-regulated anti-apoptotic genes Bcl-2 and survivin. In contrast, expression of pro-apoptotic gene Bax was up-regulated in the presence of sh-CCAT1 (Fig. 2B and C). Taken together, these data indicated that knockdown of CCAT1 enhances cisplatin-induced apoptosis via modulating the expression of apoptosis-related proteins.
3. CCAT1 acts as miR-454 sponge to down-regulate its expression.
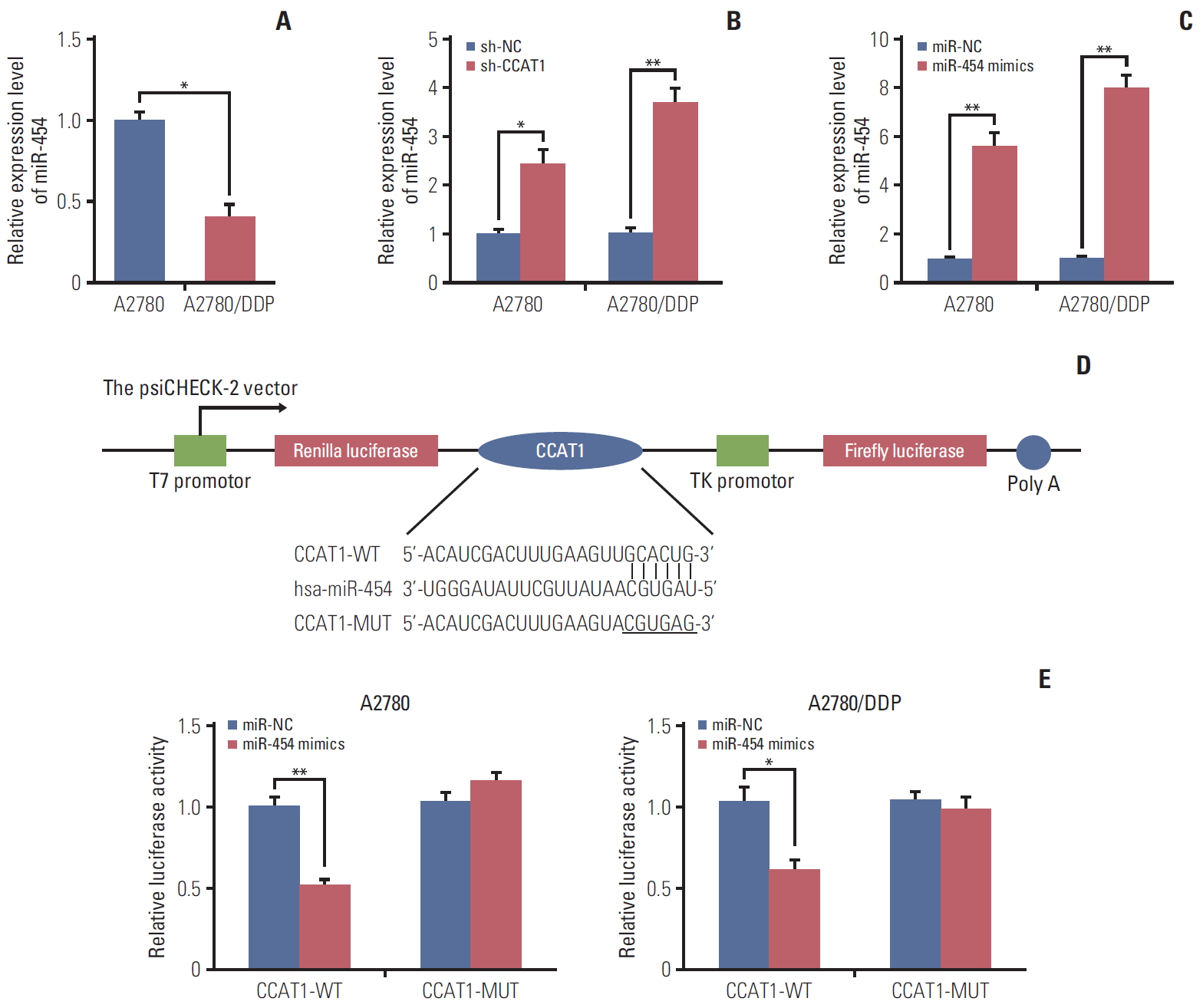
Emerging evidence has revealed that lncRNAs could act as “miRNA sponge” to bind specific miRNAs and regulate their function [14]. To identify the potential CCAT1-targeting miRNAs, online bioinformatics software Starbase (http://starbase.sysu.edu.cn/index.php) was used to search the miRNAs which have complementary base pairing with CCAT1. Among these predicted miRNAs, dramatically decreased expression of miR-454 was found in A2780/DDP cells (Fig. 3A), suggesting that miR-454 might be the target miRNA of CCAT1 in ovarian cancer cells. Furthermore, knockdown of CCAT1 caused a remarkably induction of miR-454 in both A2780 and A2780/DDP cells (Fig. 3B). The level of miR-454 was significantly up-regulated in the A2780 and A2780/DDP cells transfected with miR-454 mimics (Fig. 3C). The predicted miR-454 binding sites of CCAT1 were shown in Fig. 3D. Dual luciferase reporter assay was further performed to validate the direct interaction between miR-454 and CCAT1. As shown in Fig. 3E, co-transfection of miR-454 mimics and wild-type CCAT1 (CCAT1-WT) caused a significant reduction of luciferase activity compared with corresponding control. Conversely, mutated CCAT1 (CCAT1-MUT) almost completely abolished miR-454–caused reduction of luciferase activity (Fig. 3E). Taken together, these data suggested that CCAT1 sponges miR-454 to decrease its expression.
4. Overexpression of miR-454 modulates cisplatin sensitivity of ovarian cancer cells via promoting cell apoptosis
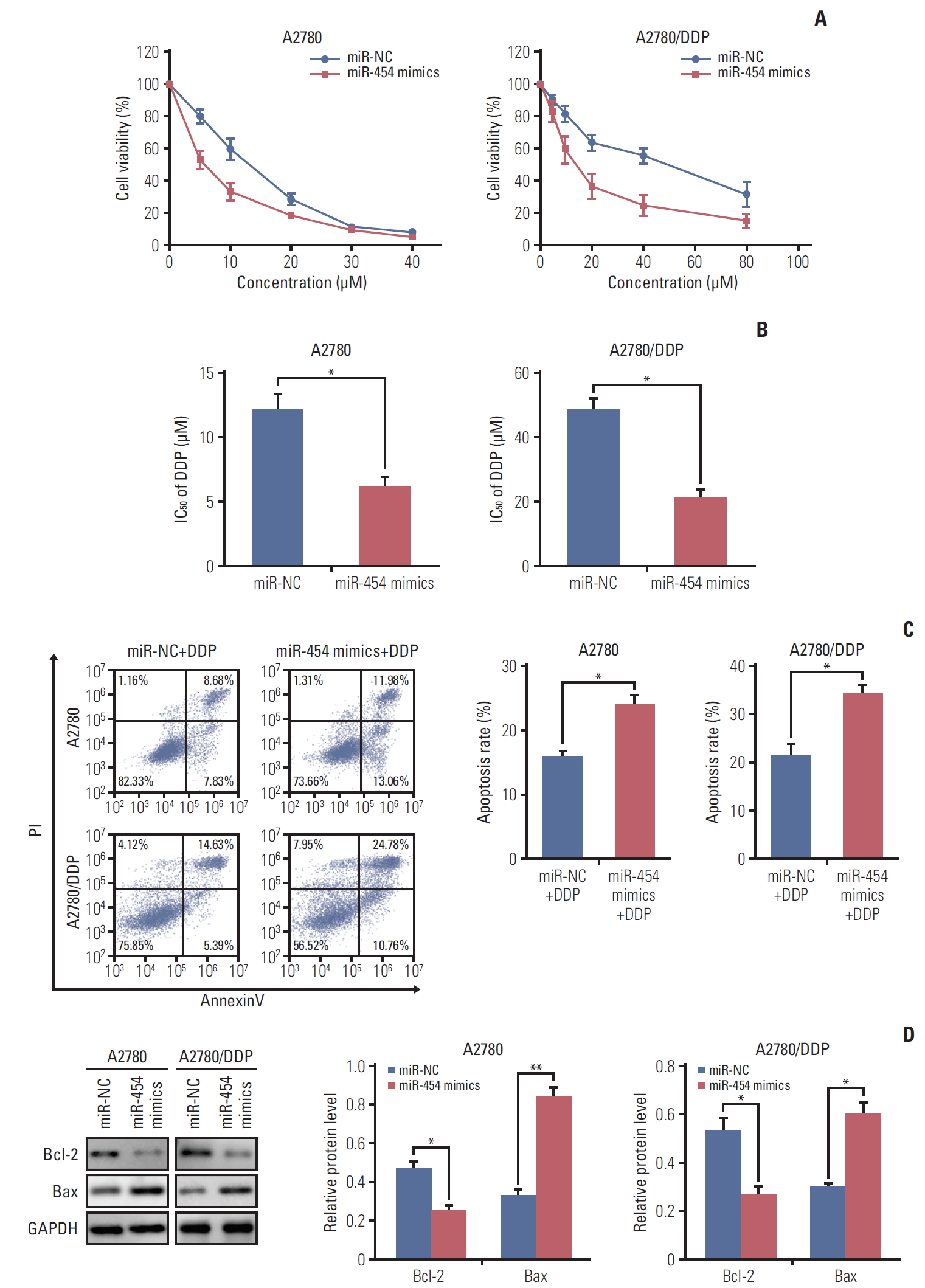
In order to unravel the effect of miR-454 on cisplatin sensitivity, MTT assays were performed in A2780 and A2780/DDP cells. As presented in Fig. 4A, overexpression of miR-454 mimics significantly enhanced sensitivity to cisplatin in both A2780 and A2780/DDP cells. The IC50 of cisplatin in miR-454 mimics transfected A2780 or A2780/DDP cells were remarkably decreased when compared with that of corresponding control cells (Fig. 4B). We further checked the effect of miR-454 on cisplatin-induced apoptosis in ovarian cancer cells. Moreover, Annexin-V-FITC/PI staining showed that overexpression of miR-454 markedly increased the apoptotic rate upon cisplatin treatment in both A2780 and A2780/DDP cells (Fig. 4C). Western blotting was further performed to detect the protein levels of apoptosis-related proteins. Consistent with the fluorescence-activated cell sorting results, overexpression of miR-454 down-regulated the protein level of anti-apoptotic Bcl-2, but up-regulated pro-apoptotic Bax (Fig. 4D). Collectively, these findings indicated that miR-454 modulates cisplatin sensitivity via promoting cell apoptosis.
5. miR-454 enhances the sensitivity to cisplatin via target of survivin in A2780 and A2780/DDP cells
We next explored the potential targets of miR-454 by using TargetScan (http://www.targetscan.org/vert_71/). Among these theoretical targets, we concentrated on the well-known apoptosis-related protein survivin given its documented role in drug resistance [15]. As presented in Fig. 5A, miR-454 binds to complementary site in the 3′-UTR of survivin. The direct interaction between miR-454 and survivin was further verified by luciferase reporter assay. A2780 or A2780/DDP cells were co-transfected with mimics negative control (miRNC)/miR-454 mimics with psiCHECK-2, psi-survivin-3’UTR-WT (survivin-WT) or psi-survivin-3′-UTR-MUT (survivin-MUT). As expected, a significant reduction of luciferase activity was observed in miR-454+survivin-WT versus miR-454+ psiCHECK-2 in both A2780 and A2780/DDP cells (Fig. 5B), whereas no obvious change was found in miR-454+survivin-MUT group. Moreover, the results suggested that overexpression of miR-454 dramatically decreased the expression of survivin at both mRNA and protein levels (Fig. 5C). Taken together, survivin is a direct target of miR-454 in A2780 and A2780/DDP cells.
To check whether miR-454 could promote apoptosis via suppressing survivin. A2780 and A2780/DDP cells were transfected with vector alone or survivin-overexpression plasmid (OE-survivin). qRT-PCR and Western blotting results confirmed that the mRNA and protein levels of survivin significantly increased after transfection (Fig. 5D). As shown in Fig. 5E, co-transfection of miR-454 mimics and survivin decreased the cell apoptotic rate after cisplatin treatment when compared with miR-454 mimics alone group. These findings suggested that overexpression of survivin attenuates the pro-apoptotic effect of miR-454 upon cisplatin treatment in ovarian cancer cells. In addition, the levels of Bcl-2 and Bax were determined by Western blotting. Consistently, survivin overexpression abolished the effect of miR-454 on Bcl-2 and Bax expression in ovarian cancer cells. Co-transfection of miR-454 mimics and survivin overexpression plasmid caused a rebound of Bcl-2 expression, but decreased Bax expression compared with miR-454 mimics alone (Fig. 5F). Collectively, miR-454 enhances the sensitivity to cisplatin via suppressing survivin in A2780 and A2780/DDP cells.
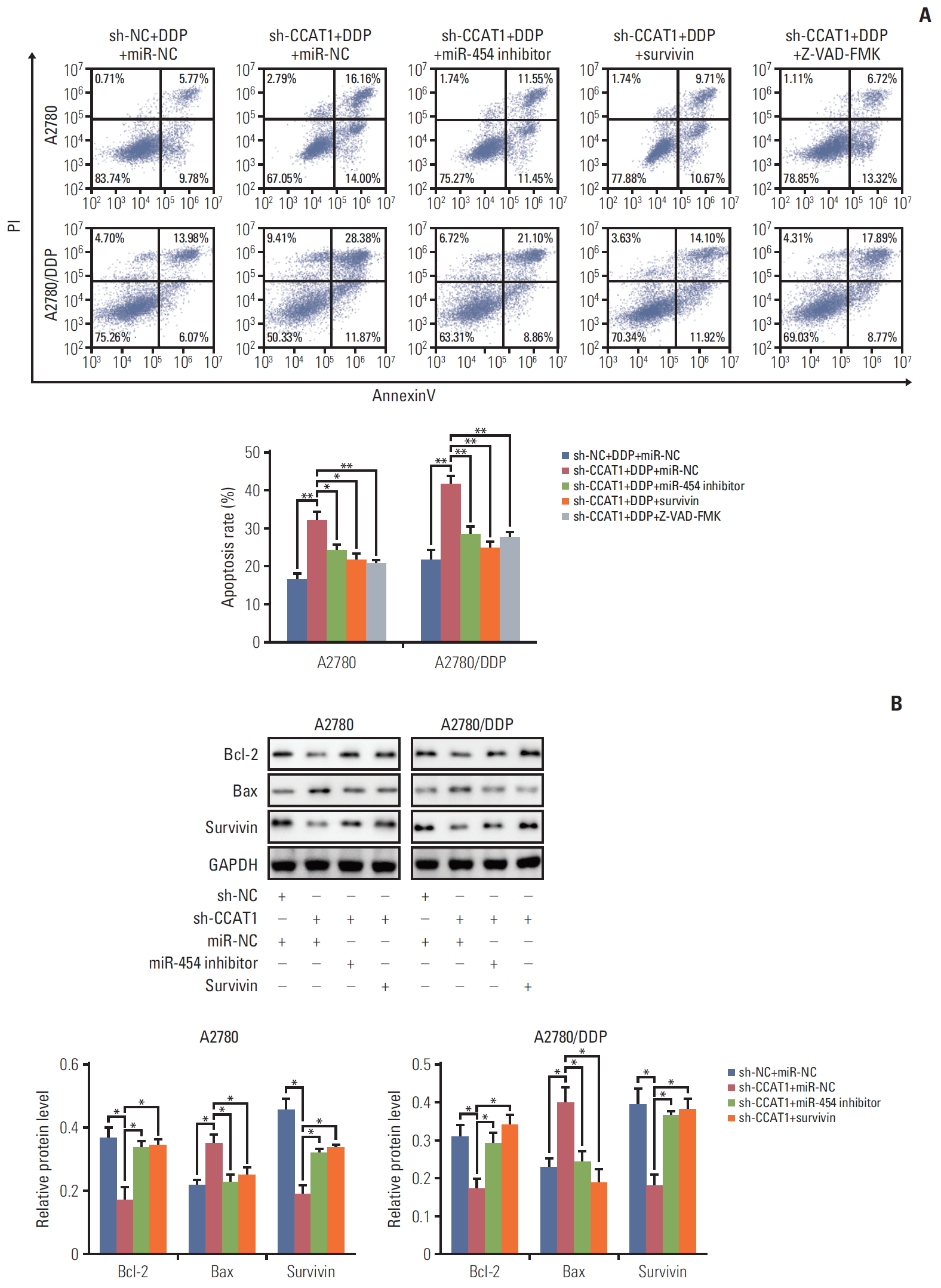
6. CCAT1/miR-454/survivin axis regulates cisplatin resistance in ovarian cancer cells
To further confirm CCAT1, miR-454 and survivin work in concert to modulate apoptosis in ovarian cancer cells, apoptosis assays were conducted. As shown in Fig. 6A, silencing of CCAT1 significantly increased the apoptotic rate upon cisplatin treatment. sh-CCAT1+miR-454 inhibitor or sh-CCAT1+ OE-survivin led to a remarkable inhibition of cisplatininduced apoptosis when compared with sh-CCAT1 group. Furthermore, a pan-caspase inhibitor of apoptosis (IAP) Z-VAD-FMK successfully inhibited the effect of sh-CCAT1, confirming that knockdown of CCAT1 induced cell death via apoptosis (Fig. 6A). The protein levels of Bcl-2, Bax and survivin were examined by western blotting. As shown in Fig. 6B, sh-CCAT1 decreased the protein levels of Bcl-2 and survivin, but increased Bax expression in both A2780 and A2780/DDP cells. In contrast, co-transfection of miR-454 inhibitor or survivin overexpression plasmid attenuated the effect of sh-CCAT1 on these apoptosis-related proteins. In addition, the role of CCAT1/miR-454/survivin axis was also validated in SKOV3 and SKOV3/DDP cells. miR-454 was down-regulated in SKOV3/DDP cells (S2A Fig.), and knockdown of CCAT1 significantly induced miR-454 or reduced survivin expression in both SKOV3 and SKOV3/DDP cells (S2B and S2C Fig.). Annexin-V-FITC/PI staining showed that CCAT1 knockdown or miR-454 overexpression potentiated cisplatin-induced apoptosis, whereas miR-454 knockdown or survivin overexpression attenuated the effect of sh-CCAT1 on apoptosis (S2D Fig.). Furthermore, knockdown of CCAT1 or overexpression of miR-454 repressed survivin and Bcl-2 expression, but induced Bax expression. miR-454 knockdown or survivin overexpression attenuated the sh-CCAT1-mediated dysregulation on survivin, Bcl-2 and Bax levels (S2E Fig.), suggesting that CCAT1 sponges miR-454 to regulate the expression of Bcl-2, Bax, and survivin in ovarian cancer cells.
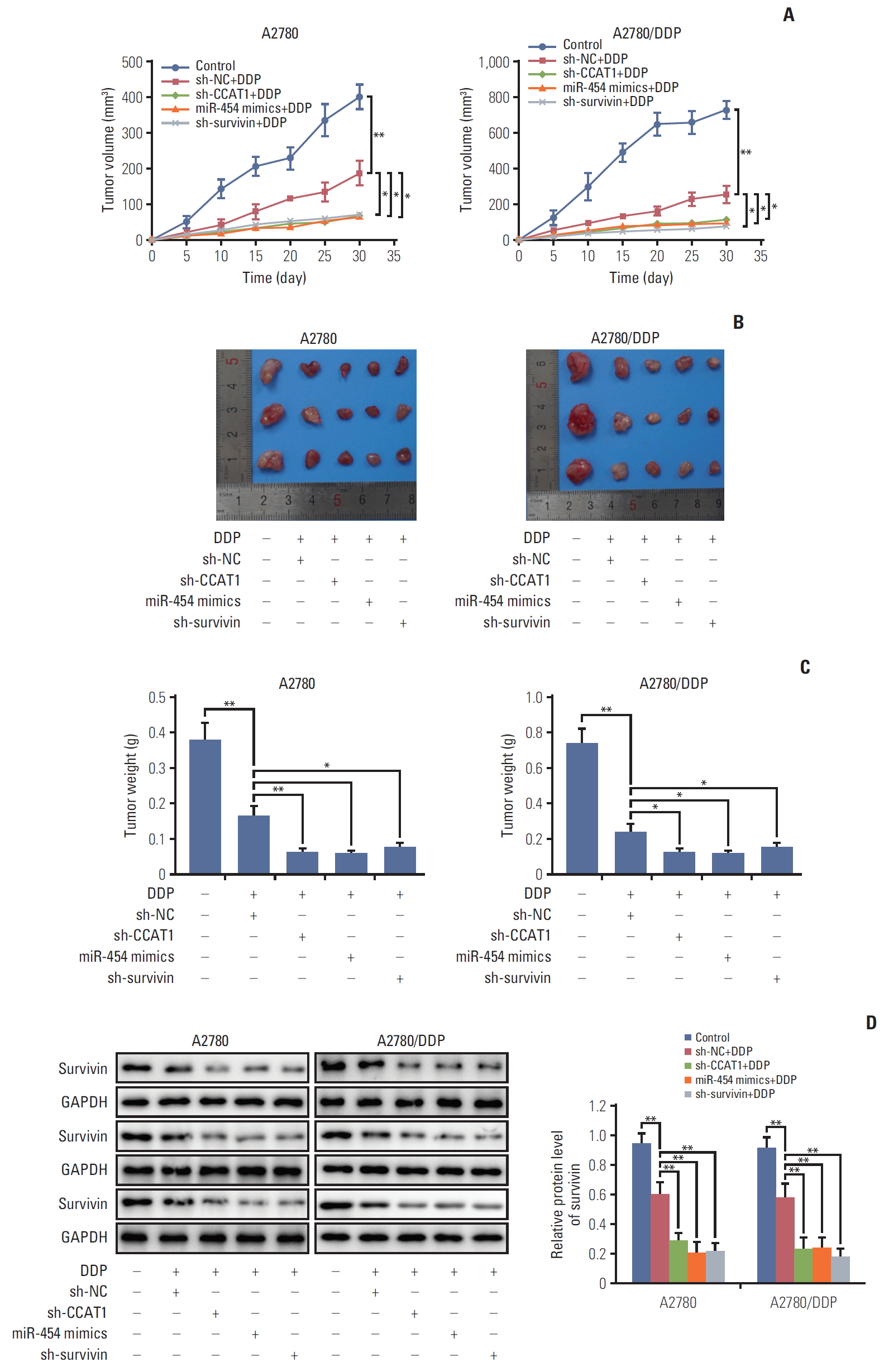
7. Knockdown of CCAT1 enhances sensitivity to cisplatin in vivo
To further assess the effect of CCAT1/miR-454/survivin on cisplatin sensitivity in vivo, we characterized tumor formation and progression in xenograft model by assessing tumor volume and weight. shRNA NC, miR-NC, miR-454 mimics, sh-CCAT1, or sh-survivin were transfected into A2780 and A2780/DDP cells. Nude mice were then inoculated with these cells and treated with vehicle control or cisplatin. It is worthy to note that mice in A2780 and A2780/DDP groups received 5 mg/kg and 10 mg/kg cisplatin, respectively. A2780/DDP cells were more resistant to cisplatin treatment, therefore, mice in this group were treated with a higher concentration of cisplatin (10 mg/kg). To assess whether tumor growth was affected by CCAT1, subcutaneous tumor volume was measured over time. Tumors derived from CCAT1 depleting A2780 or A2780/DDP cells grew more slowly than those formed by NC transfected cells upon cisplatin treatment (Fig. 7A and B). Interestingly, similar results were observed in miR-454 mimics+ DDP and sh-survivin+DDP groups (Fig. 7A and B). On day 30 following treatment, tumors were collected and weighed. As shown in Fig. 7C, knockdown of CCAT1 or survivin and overexpression of miR-454 mimics caused a more significant reduction of tumor weight upon cisplatin treatment in both A2780 and A2780/DDP cells compared with corresponding controls. These findings further address the role of CCAT1/miR-454/survivin axis on chemoresistance to cisplatin in vivo. Down-regulation of survivin protein was observed in sh-CCAT1+cisplatin, miR-454 mimics+cisplatin and sh-survivin+cisplatin tumors when compared with NC+cisplatin tumors (Fig. 7D). Taken together, these data suggested that deregulated CCAT1/miR-454/survivin axis is implicated with cisplatin resistance in ovarian cancer.
Discussion
Ovarian cancer is the most fatal tumor of female reproductive system. The conventional treatment is severely limited by chemoresistance [16]. The thorough understanding of the resistance and underlying mechanism of EOC progression is urgently needed. CCAT1 has been previously proposed to be associated with high metastatic potential in ovarian cancer cells [11,17]. In this study, we demonstrated that CCAT1 was significantly up-regulated in cisplatin-resistant A2780/DDP cells, indicating that CCAT1 is also implicated in cisplatin resistance in ovarian cancer cells. Knockdown studies suggest that lack of CCAT1 enhanced sensitivity to cisplatin in which cisplatin-induced apoptotic rate significantly increased in CCAT1 depleting A2780 or A2780/DDP cells. Bioinformatic analysis coupled with dual luciferase assay revealed that CCAT1 served as miR-454 sponge, and survivin was identified as a functional target of miR-454. Moreover, gain and loss experiments showed that CCAT1/miR-454/survivin axis contributed to cisplatin resistance by promoting apoptosis in A2780 and A2780/DDP cells. These in vitro data were confirmed by xenograft study in which the mean volume and weight of tumors derived from sh-CCAT1+cisplatin cells were much smaller than that of control cells.
A number of studies have demonstrated that CCAT1 plays a crucial role in drug resistance in different cancer cells. For instance, CCAT1 exerts oncogenic role and promotes drug resistance in docetaxel-resistant lung cancer cells [7]. It is well-established that chemotherapy agents induce cancer cell death, at least in part, through apoptosis [18]. In ovarian cancer, cisplatin is the most frequently used chemotherapy agent [19]. Dysregulation of key apoptotic factors, including the IAP family, p53, Akt, and death-receptor family, is critical for cisplatin resistance in ovarian cancer cells [20]. However, whether CCAT1 contributes to cisplatin resistance via modulating these key apoptotic factors remains unexplored. Here, we revealed that knockdown of CCAT1 significantly decreased the viability and promoted apoptosis of ovarian cancer cells after cisplatin treatment. Silencing of CCAT1 also caused a remarkable reduction of anti-apoptotic Bcl-2 and survivin, but an induction of pro-apoptotic Bax, indicating that these apoptosis-related proteins might function as downstream effectors of CCAT1 in response to cisplatin in ovarian cancer cells. These findings suggested that CCAT1 might be a promising therapeutic target for chemotherapy of EOC.
Accumulating evidence indicates that lncRNAs act as sponges for miRNAs and abrogate the suppressive effect of miRNAs on their target gene [14]. CCAT1 also plays a critical role in the ceRNA network to regulate drug resistance. Chen et al. have demonstrated that CCAT1 promotes chemoresistance by sponging let-7c [7]. A more recent study has reported that CCAT1/miR-130a-3p/SOX4 axis increases cisplatin-resistance in non-small-cell lung cancer [21]. Although bioinformatics analysis has predicted that CCAT1 harbored the recognition sequence of many miRNAs, we focused on miR-454 because of its low expression in cisplatin resistant A2780/DDP cells. In recent years, dysregulated miR-454 expression was found in variety types of cancers. It functions as oncogene or tumor suppressor in different tumor types. For instance, miR-454 promotes colorectal cancer cells (CRC) proliferation by targeting CYLD [22]. The oncogenic role of miR-454 was also found in HCC and triple negative breast cancer (TNBC), and miR-454 is associated with poor prognosis in HCC or TNBC patients [23,24]. By contrast, miR-454 inhibited cell proliferation and invasion, but induced apoptosis in gastric cancer (GC) cells by targeting MAPK1 [25], illustrating its tumor suppressive role in GC. Importantly, a more recent study has illustrated that lncRNA HOXA11-AS acts as ceRNA of miR-454 to promote cisplatin resistance in lung adenocarcinoma (LUAD) cells [26]. In the current study, we demonstrate for the first time that miR-454 plays a crucial role in cisplatin resistance in ovarian cancer cells. miR-454 was identified as a direct target of CCAT1 in ovarian cancer cells. Overexpression of miR-454 promoted cisplatin-induced apoptosis in both A2780 and A2780/DDP cells, whereas miR-454 inhibitor attenuated the effect of CCAT1 on apoptosis, suggesting that CCAT1 promoted cisplatin-induced apoptosis via sponging miR-454.
Survivin (also known as BIRC5), the smallest member of IAP family, functions as an anti-apoptotic proteins through inhibition of caspase [27]. Emerging evidence suggests that survivin is associated drug-resistance in ovarian cancer cells. For instance, dihydromyricetin restore drug sensitivity in ovarian cancer by p53-induced down-regulation of surviving [28]. A more recent study has reported that Smac reverses drug resistance of ovarian cancer cells via inhibiting surviving [29]. Given its critical role in chemoresistance, we thus test whether survivin acted as a functional target of miR-454 in ovarian cancer cells. As expected, bioinformatic analysis and luciferase reporter assay indicated that miR-454 directly associated with 3′-UTR of survivin and suppressed its expression. Overexpression of miR-454 mimics potentiated cisplatin-induced apoptosis through modulating Bcl-2 and Bax, whereas enforced expression of ectopic survivin partially attenuated the effect of miR-454 on cell apoptosis. Overexpression of survivin also abolished the effect of sh-CCAT1 on apoptosis, suggesting a crucial role of survivin in CCAT1/miR-454 ceRNA signaling. The xenograft results confirmed that CCAT1 acted as a modulator of cisplatin response via miR-454/survivin in vivo. Our data revealed that CCAT1 regulated survivin by competitively sponging miR-454, and thus conferred cisplatin resistance of ovarian cancer cells. Targeting CCAT1/miR-454/survivin axis may provide a promising strategy for reversing cisplatin resistance in ovarian cancer cells.
In conclusion, we first demonstrated that CCAT1/miR-454/survivin axis contributed to cisplatin resistance in ovarian cancer cells. This study provides better understand of molecular mechanism of CCAT1 in chemoresistance of EOC.