Introduction
Pineal region tumors (PRTs) are a heterogeneous group of tumors that can be assigned to four main categories: germ cell tumors, pineal parenchymal tumors (PPTs), glial cell tumors, and other miscellaneous tumors and cysts. PRTs, which are rare tumors, account for approximately 0.4-1.0% of intracranial tumors. PPTs are even rarer, and approximately 30% of PPTs occur in neoplasms of the pineal region [1].
According to the World Health Organization (WHO) classification of tumors in the central nervous system, which was revised in 2007, PPTs are subdivided into well differentiated pineocytoma (PC), PPT with intermediate differentiation (PPTID), and poorly differentiated pineoblastoma (PB). A standard treatment strategy for these tumors has not yet been established. Various treatments from surgery or radiotherapy and chemotherapy alone or in combination have been applied. Here, we report on a case of effective treatment of PPTID. Part of the tumor was initially removed through surgery followed by radiation therapy; however, this treatment was not effective; therefore, chemotherapy was applied and resulted in successful elimination of the tumor.
In this study, we also discuss the pathological and biological features of PPT and available treatment options.
Case Report
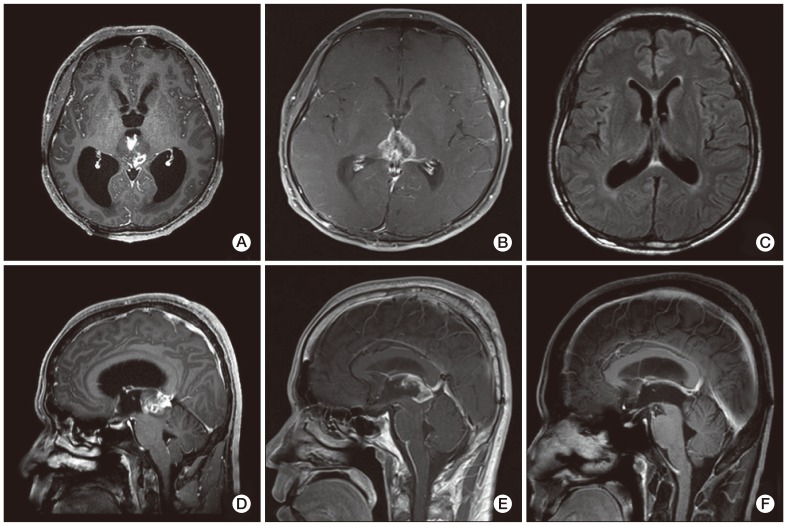
A 37-year-old male visited a clinic due to a headache and dizziness in December 2010. There was no distinguishable observation in his past history, physical examination, and laboratory tests. In addition, the head-up-tilt-table test and video eyeball exam showed no obvious abnormalities. Computed tomography (CT) of his brain was performed and a mass measuring 2.5 cm in size was observed in the midbrain and obstructive hydrocephalus. The tumor showed intermediate enhancement in T1-weighted magnetic resonance (MR) imaging and a high intensity of enhancement in T2-weighted MR (Fig. 1A and D). Differential diagnosis based on MR imaging included germinoma, PPT, and glial tumor. Neurosurgeons performed an immediate occipital craniotomy and partial removal of the tumor in 2010. Histological diagnosis concluded that the mass was a PPT with intermediate differentiation. The majority of the tumor tissue consisted of small hyperchromatic cells with moderately increased cellularity and a high nuclear to cytoplasmic ratio. The tumor cells showed some atypical mitosis but rarely exhibited mitotic activity. A clear Homer Wright rosette, many of which have been termed "pineocytomatous resettes" was not found (Fig. 2B). Diffuse staining for neuron specific enolase and focal staining for synaptophysin were observed, while the immunohistochemistry analysis showed very little glial fibrillary acidic protein (GFAP) staining. Such a focal staining pattern of synaptophysin staining in the pineal region neoplasm, coupled with the lack of GFAP staining was most consistent with a PPT (Fig. 2A).
He underwent irradiation with 54 Gy of radiation on 27 fractions for removal of the remaining tumor for approximately one month after surgery. However, a remnant mass in the superoposterior aspect of the midbrain had extended to the hypothalamus and the third ventricle approximately five months after the operation. MR imaging showed leptomeningeal enhancement in the infratentorial fossa, indicating tumor cell seeding. (Fig. 1B and E). He was referred to the department of medical oncology for palliative chemotherapy and was treated with a procarbazine, lomustine, vincristine (PCV) regimen one month later. The size of the remaining tumor in the midbrain and the leptomeningeal enhancement was reduced in the follow-up brain MR imaging. Two months later, after the sixth cycle of PCV chemotherapy, no evidence of the tumor was observed in the brain MR imaging (Fig. 1C and F). No additional signs of a tumor were observed in the latest follow-up imaging, which was performed approximately 20 months after the initial diagnosis. The patient was free of any signs of recurrence without any neurological deficits six months after the end of treatment.
Discussion
PRTs are derived from cells located in and around the pineal gland. The most common PRTs are germ cell tumors and PPTs. There are still some controversies with regard to PPT classification, primarily due to the rare occurrence of these tumors. Many older published studies (prior to 2007) separated PPT into only two histological subtypes; PC and PB. PC was classified as an indolent tumor and designated as grade I with a benign feature. PB was categorized as a highly malignant primitive neuroectodermal tumor and assigned a WHO grade IV, which tends to have a higher propensity for seeding the cerebrospinal axis.
The 2007 WHO classification of tumors of the central nervous system designated a new category of PPT, called PPTID, which describes a group of tumors that have an intermediate malignancy between PC and PB. Jouvet et al. [2] proposed a new prognostic grading system that subdivides the category of PPTID into two groups with different prognoses (grades II and III) according to the mitotic activity and the degree of neuronal differentiation. Grade III has been defined as PPTID with either six or more than six mitoses or fewer than six mitoses but no immunostaining for neurofilaments. In our case, the patient had been diagnosed as PPTID grade III, since the tumor contained more than six mitoses and atypical mitosis. Fauchon et al. [3] reported that grade III PPTID had a much more aggressive biologic behavior compared with grade II. They reported that grade II and grade III differed by 74% and 39% in five-year survival and by 26% and 56% in the recurrence rate, respectively.
A previous report compared the histological types between initial diagnosed and recurrent tumors in PPTID. A 47-year-old male who had been diagnosed with PPTID III through endoscopic third ventriculostomy was treated with gamma knife surgery and no residual tumor was observed. However, a new mass lesion appeared on the cerebellum four years later and was found to be PB after a creniectomy and open biopsy were performed. In regard to the reoccurrence of PPT, no cases of a changed diagnosis have been reported, except for the report described above. However, PPT might be morphologically heterogeneous within a tumor and an endoscopic biopsy might not be sufficient for observation of the whole tumor. The authors even stated that the first tumor might have been a mixed PPTID-PB [4]. Although we did not perform a repeat biopsy in our case, it is likely that the recurred cancer was also a PPTID of high grade.
The definitive management of these tumors remains unclear. A previous study recommended a variety of treatment approaches ranging from surgery or external irradiation alone to combined treatment with surgery, radiotherapy, or chemotherapy [5].
Surgery has been reported to play a significant role in relieving the local mass effect and providing a maximal tissue sample for histological analysis of low-grade PPT. Despite the significant progress in surgical techniques and perioperative care, surgical intervention in the pineal area is high risk because of its location near critical structures. Contemporary studies have reported a surgical mortality rate of 4-7%, and the permanent morbidity rate may be as high as 10% [6]. Some authors found no correlation between the extent of resection and survival, although total resection showed an association with better local control. In a study of a children's cancer group, which included 25 PRT patients, no significant difference in survival was observed (<90% surgical resection vs.≥90% surgical resection (p>0.3, data not shown). This was likely due to the small sample size [1].
Another treatment option for PPT patients is radiotherapy and chemotherapy. PCs are treated with surgical resection followed by conventional local radiotherapy or stereotactic radiosurgery. PBs or high grade PPTIDs usually require more aggressive treatment, including craniospinal irradiation and chemotherapy. A retrospective study of 22 PPT patients with PB, mixed PPT or PPTID with seeding potential found that fractionated irradiation applied primarily or as an adjuvant postoperatively could control the tumor and increase survival. The one-year, three-year, and five-year survival rates for these patients (seeding varieties) were 88%, 78%, and 58% [7].
The indications and protocols were heterogenous in the initial stages when chemotherapy was administered during postoperative treatment. Because of the harmful effect of radiotherapy on the developing nervous system and the hypothalamic-pituitary axis in children, concerns about radiation toxicity have prompted efforts to reduce radiation therapy. In the Pediatric Oncology Group (POG) experience, all 11 reported children under the age of 36 months with PB were treated with multi-agent chemotherapy for 12-24 months in order to delay radiation until the age of 36 months. They ultimately failed chemotherapy and all died of disease [8]. Gururangan et al. [9] studied the efficacy of high-dose chemotherapy with or without craniospinal radiotherapy for PB. They reported that five of 12 patients who had undergone radical tumor resection followed by chemotherapy survived and four of seven patients treated with chemotherapy after biopsy or incomplete resection lived without any recurrences for a median follow-up period of 62 months. Recent studies on chemotherapy have focused primarily on PB and few studies on PPTID have been reported [10,11]. Table 1 shows the various chemotherapy options that have been reported. Kurisaka et al. [11] reported on a patient with PPTID II who had received treatment with 36 Gy of craniospinal irradiation and an additional local boost of 19.8 Gy after undergoing partial tumor resection. Positron emission CT after surgery and irradiation indicated the presence of a residual tumor. The patient received one cycle of intensive chemo-therapy consisting of cisplatin, oral etoposide, cyclophosphamide, and vincristine. Chemotherapy could not be continued due to development of thrombocytopenia, and cyberknife radiotherapy (marginal doses of 23 Gy/3 fractions) was performed two months later. The report stated that the patient was free of any signs of recurrence without any neurological deficits 17 months after the end of cyberknife radiotherapy [5].
In this study, we report on a case of PPTID, in which remission was achieved through PCV chemotherapy. PPTID is extremely rare and few large-scale studies have reviewed the long-term effects of treatment, and there have been no definite conclusions regarding treatment plans for PPTID in adults. In addition, few cases involving successful treatment of PPTID in adults with neoadjuvant chemotherapy and craniospinal irradiation have been reported. We would like this form of treatment to help a larger number of PPT patients. Therefore, based on the findings of this case, combining surgery and irradiation with chemotherapy may be a feasible and efficient therapeutic approach for treatment of PPTID. However, conduct of a prospective study including a larger number of patient groups will be needed in order to conclusively demonstrate the effectiveness of chemo- and radiotherapy for treatment of PPTs.