Introduction
Erdheim-Chester disease (ECD) is a rare non-Langerhans-cell histiocytosis of unknown etiology involving infiltration of bones and various organs by lipid-laden histiocytes with foamy or eosinophilic cytoplasm. The histiocytes are CD68-positive and CD1a-/S100-negative. Lipid-laden histiocytes produce xanthogranulomas, which can be found in long tubular bones, skin, lung, heart, kidney, retroperitoneal space, orbit, and pituitary gland.
ECD primarily affects adults. The mean age of the diseased population is 53 years [1], and there is a slight predominance of males. Most patients have skeletal involvement at the time of diagnosis; extra-skeletal involvement occurs in approximately 50% of patients. The bone involvement is constant and includes symmetric osteosclerosis of long bones, especially of the low extremities. ECD rarely infiltrates the axial skeleton and lymph nodes [2].
First described by Erdheim and Chester in 1930 [1], fewer than 500 cases of ECD have been reported in the published literature. In Korea, the first case of ECD was described in 1999 [3], and 15 cases have since been reported. Herein, we report on a unique case of ECD with bone marrow, axial skeleton, and lymph node involvement.
Case Report
In March 2009, a 76-year-old man with a 20-year history of hypertension was referred to SMG-SNU Boramae Medical Center with exertional dyspnea. Despite treatment with antihypertensive medication, his blood pressure was not well controlled. Physical examination of the patient revealed mild splenomegaly. White blood cell, hemoglobin, and platelet counts were 3,180 cell/mm3, 7.4 g/dL, and 65,000/mm3, respectively. There was no albumin/globulin ratio reversal.
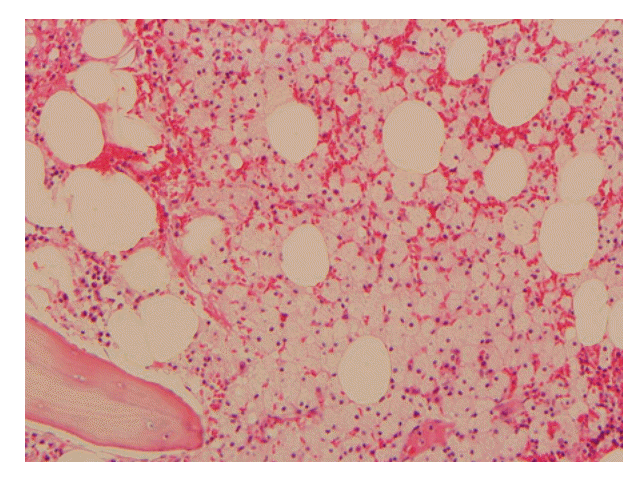
Serum and urine protein electrophoresis, immunoelectrophoresis, and immunofixation electrophoresis of serum were normal. Serum blood urea nitrogen and creatinine levels were 43 and 2.3 mg/dL, respectively. There was heavy proteinuria in 24-hour urine protein, and his 24-hour urine protein was 1,816 mg/day. Thyroid-stimulating hormone and free thyroxine levels were 16.63 μIU/mL and 0.71 ng/mL, respectively. Chest radiograph was normal. Small-sized kidneys and mild splenomegaly (13.2 cm) were observed by abdominal ultrasonography. Cardiac contraction was normal with an ejection fraction of 61.8% and a pattern of mild diastolic dysfunction was observed by echocardiography. Bone marrow examination showed cellularity of 60%, which was slightly hypercellular for the patient’s age, with slightly increased foamy histiocytosis (Fig. 1). Immunohistochemical stains were negative for acid-fast bacilli, Oil Red O, and periodic acid Schiff, eliminating the likelihood of infectious causes. The serum iron profile was suggestive of an anemia of chronic disease. We diagnosed the patient with anemia of chronic disease, cytopenia related to chronic illness, chronic renal failure due to hypertension, and hypothyroidism. However, we could not determine a definite cause or explanation for the cytopenia. We initiated treatment with levothyroxine for the hypothyroidism.
In September 2009, the patient was admitted with exertional dyspnea and generalized edema. Pretibial edema and arrhythmia were observed. The patient’s complete blood count was still compatible with anemia of chronic disease accompanied by mild thrombocytopenia, which was not significantly different from what was observed at the initial visit 6 months earlier. Electrocardiography showed atrial fibrillation. Echocardiography showed normal systolic function, with an ejection fraction of 67%, and mild diastolic dysfunction, which was not different from what had been observed previously. Coronary angiography showed 50% stenosis in the left anterior descending branch of the left coronary artery and right coronary artery. The patient was treated with diuretics, which resulted in improvement of the dyspnea and generalized edema. The patient was discharged without coronary intervention and with plans to continue medical therapy and follow-up in the nephrology and cardiology departments.
In May 2011, the patient presented with progressive dyspnea and generalized edema, without additional symptoms. White blood cell, hemoglobin, and platelet counts were 5,190 cell/mm3, 7.2 g/dL, and 72,000/mm3. Serum blood urea nitrogen and creatinine levels were 92 mg/dL and 6.4 mg/dL, respectively. Serum potassium levels were elevated to 7.6 mEq/L. Liver function tests and thyroid function tests were normal. When the bone marrow examination was repeated, cellularity was slightly decreased and histiocyte infiltration was increased when compared to the previous examination. The patient’s renal function had deteriorated to chronic renal failure stage V. Hemodialysis was initiated, and the patient’s symptoms improved.
In January 2014, the patient presented with low back pain that had originally started 2 years earlier and had recently become aggravated. On physical examination, the patient had multiple enlarged lymph nodes in both axillary areas and did not show any signs of fever or weight loss. Complete blood count and peripheral blood smear revealed normocytic normochromic anemia and mild thrombocytopenia. The results of liver function tests, lipid profiles, and tumor markers (α-fetoprotein, 1.66 ng/mL; carcinoembryonic antigen, 1.82 ng/mL; cancer antigen [CA]-19-9, 3.44 U/mL; CA-125, 10.7 U/mL) were normal. There was no albumin/globulin ratio reversal. Urine test for Bence-Jones protein was negative. Serum and urine protein electrophoresis was normal. Immunoelectrophoresis and immunofixation electrophoresis of serum were normal. Serum free light chain was also normal.
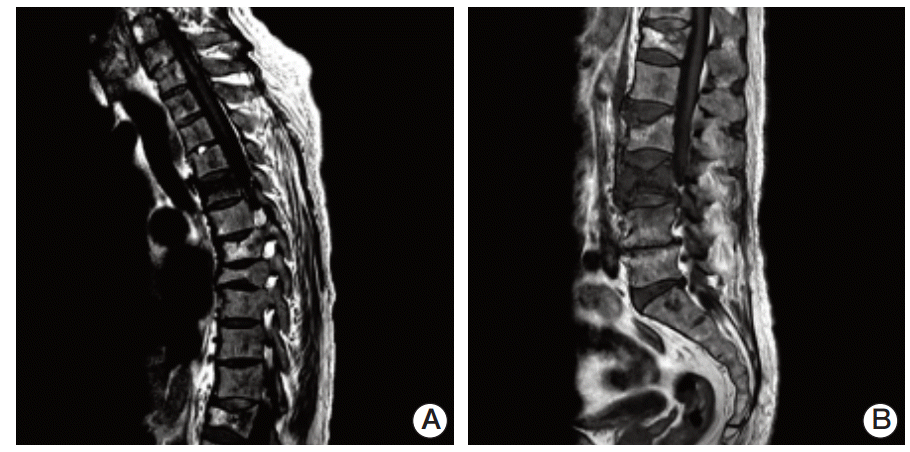
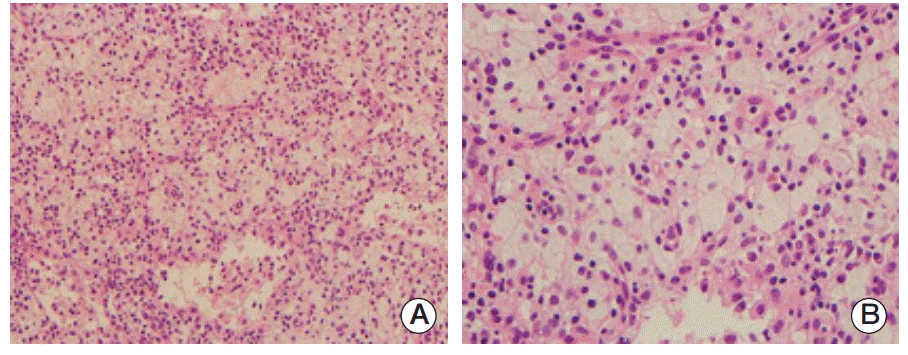
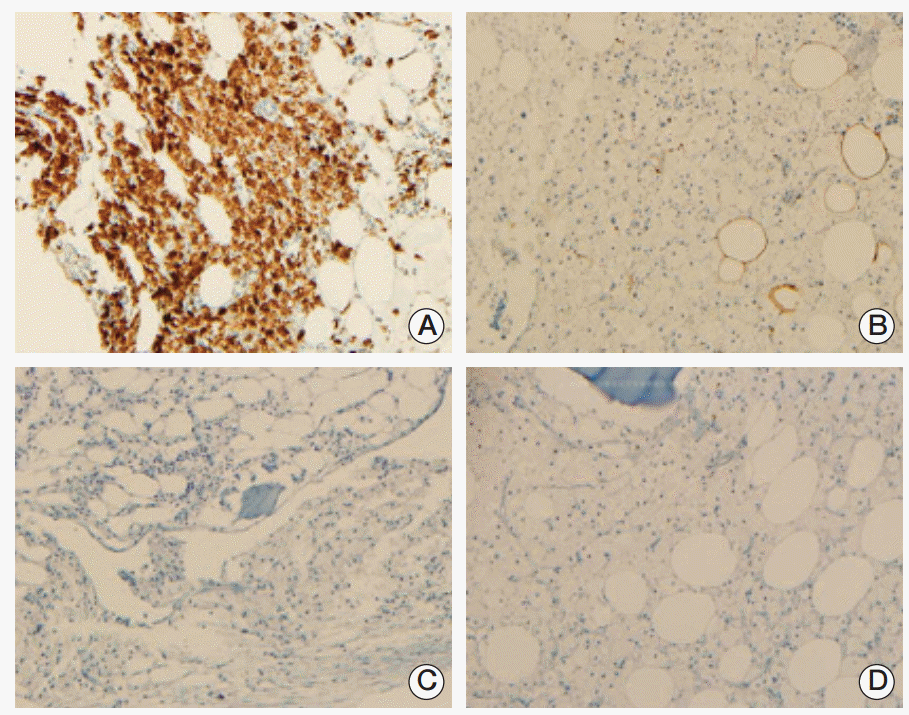
Compression fractures of the T5, L3, and L4 vertebral bodies were identified by magnetic resonance imaging of the spine (Fig. 2A and B). In particular, the L3 vertebral body showed extensive bone marrow edema and paravertebral soft tissue swelling, with an anterior bulging contour suggestive of a pathologic etiology such as myeloma or metastases. Involvement of the metaphysis and diaphysis, with relative sparing of the epiphysis, was also observed. 18F-Fluorodeoxyglucose positron emission tomography showed diffuse hypermetabolic activity in the T5 and L3 vertebral bodies, the right proximal humerus, and both axillary lymph nodes. Technetium-99m bone scintigraphy showed increased uptake in T5-6, T7-8, T12, and L3-5 and both tibia. Plain radiography of the humerus showed sclerotic lesions in both anatomical neck areas of the humerus. Compression fractures were observed by a computed tomography (CT) scan of the patient’s chest in T5, T7-8, T12, and L3-4 spines. In addition, CT imaging identified cardiomegaly and multiple prominent lymph nodes in both axillae. Irregular sclerotic change was observed in both femur necks (Fig. 3). There were no significant focal lesions in the lung parenchyma. Magnetic resonance imaging of the brain showed unremarkable findings. The CT scan of the abdomen was unremarkable, other than the observation of relatively small sized kidneys and mild splenomegaly (14.4 cm). The degree of splenomegaly was unchanged compared with previous studies. Bone biopsy of the L3 vertebral body and lymph node biopsies were performed. Histopathological examination of the bone biopsy revealed marrow fibrosis and granulated tissue, with infiltration of lipid-laden histiocytes. There was no evidence of malignancy or specific infection. Axillary lymph node biopsy revealed diffuse infiltration of lipid-laden histiocytes (Fig. 4), which were found to be positive for CD68 and negative for S100 and CD1a. There were no mutations in BRAF V600E mutation-specific antibody stains of the bone and the axillary lymph node. In addition, mutation V600E of BRAF was not detected by direct DNA sequencing of BRAF gene exon 15. Bone marrow biopsy was also performed, which showed the same results as the bone and axillary lymph node biopsy. With the suspicion that bone marrow involvement of ECD had been present since March 2009, we reviewed the results of the bone marrow biopsy performed in March 2009. Immunohistochemical stain of the bone marrow biopsy examined in March 2009 showed that the infiltrating histiocytes were positive for CD68 (Fig. 5A) and negative for S100, CD1a (Fig. 5B and C), and BRAF (Fig. 5D).
Based on the radiological and histopathological findings, a diagnosis of ECD with involvement of the bone, bone marrow, and axillary lymph nodes was made. We also suspected ECD involvement of the heart and kidney; however, this could not be accurately confirmed without tissue analysis. The patient was administered prednisolone; however, the treatment was not effective.
Discussion
ECD is a rare, non-Langerhans cell, histiocytic, infiltrative disorder primarily involving the peripheral long bones. It can also involve other organ systems, including the musculoskeletal, central nervous, cardiac, pulmonary, and renal systems. The pathological features of ECD are diffuse xanthogranulomatous infiltration of large foamy histiocytes, rare Touton-like giant cells, and medullary fibrosis. In immunohistochemical staining, the tumor cells stain positive for CD68 and negative for CD1a and S100.
The most commonly involved bones in ECD are the femur, tibia, and fibula; the ulna, radius, and humerus are less frequently involved. ECD involved bones are characterized by bilateral and symmetric sclerosis of the metaphyseal regions of the long bones, while the epiphysis is spared. Approximately half of all patients have extra-skeletal manifestations, which can include retroperitoneal fibrosis, orbital infiltration, interstitial lung disease [4], bilateral adrenal involvement [5], testis infiltration [6], and involvement of the breast, the central nervous system [7], or cardiovascular systems [8]. The clinical presentation of ECD ranges from asymptomatic to various and multiple symptoms related to the involved organs. The clinical details of the largest number of reported cases to date were described retrospectively by Veyssier-Belot et al. [2] and included 52 previously reported cases and seven additional cases. In this report, patients with ECD most commonly presented with juxtaarticular bone pain, usually of the knees and ankles.
It is very unusual for the lymph nodes, liver, spleen, or axial skeletons to be affected in ECD [3]; however, these areas are frequently affected in Langerhans cell histiocytosis and Rosai-Dorfman disease (RDD). In the case presented herein, the patient presented with an unusual pattern of involvement of bone marrow, axial skeleton, and lymph nodes. Involvement of lymph nodes was first documented by Sheu et al. [6] in a report on a 50-year-old white male who presented with hypogonadism and diabetes insipidus.
Because the clinical characteristics of ECD are largely unknown and it is difficult to diagnose, it is important to be clinically critical. Diagnosis of ECD is based on the pathologic evaluation of the involved tissue in combination with the clinical context. Several differential diagnoses are generally possible based on the presenting features, resulting in the necessity of a battery of investigations. It is important to consider a number of differential diagnoses, including metabolic and endocrine disorders, malignancies, multiple sclerosis, and thyroid abnormalities.
The primary differential diagnosis should include Langerhans cell histiocytosis. Langerhans cell histiocytosis presents with an infiltrate of histiocytes, which have a characteristic reniform nuclear morphology and express the markers of Langerhans cells, including CD1a, S100, and Langerin. In contrast, ECD tumor cells lack central nuclear grooves and Birbeck granules that are typical of Langerhans cell histiocytosis. In addition, ECD cells do not express CD1a or S-100 [9]. The primary differential diagnosis should also include RDD, which presents as a benign disorder with histiocyte accumulation in the lymph nodes and occasionally in the skin. The pathology of RDD is typified by prominent emperipolesis and a mixed infiltrate with histiocytes that express S100.
ECD tumor cells have a similar immunohistochemical staining pattern to juvenile xanthogranuloma (JXG) cells. JXG cells express factors VIIIa and CD68 and are negative for CD1a, Langerin, and S100. However, the clinical presentation and age of our patient were not compatible with JXG. Systemic JXG occurs mainly in young children [10], whereas ECD is a disease of middle-aged and elderly adults, with a mean age at diagnosis of 53 years (range, 21 to 77 years). JXG is usually benign and self-limiting and bone involvement is more likely to be focal, and lytic. In contrast, ECD is often a fatal disorder, with death occurring in more than 50% of patients. Death often occurs within one year of diagnosis. Approximately 60% of patients died of ECD within 3 years from initial presentation [2]. Prognosis is worse in patients with extra-skeletal involvement [2]. Death often occurs as a result of cardiac or respiratory failure resulting from heart or lung involvement.
Optimal treatment for ECD has yet to be established. Systemic steroids, various cytotoxic agents [2], radiation therapy, and autologous stem cell transplantation have all been used in treatment of patients with this condition [11]. Glucocorticoids are reserved for patients who cannot tolerate more aggressive systemic therapies and for patients with very mild symptoms who wish to avoid the potential side effects associated with more aggressive therapies. Radiotherapy may be used for local palliation. A recent study reported that interferon-α is a valuable therapy in treatment of ECD patients [12]. If interferon-α fails, imatinib mesylate has been suggested as an experimental alternative therapeutic regimen by some investigators [13]. In addition, it has been suggested that treatment with a BRAF inhibitor could be a useful therapy, as patients with multisystemic and refractory ECD and the BRAF V600E gene mutations have demonstrated partial clinical responses following treatment with the BRAF inhibitor [14].
In summary, our patient underwent a bone marrow examination for diagnosis in March 2009 because of cytopenia in the peripheral blood. Although bone marrow examination revealed increased foamy histiocytosis, we failed to diagnose ECD initially because there was no bone lesion or any other symptoms that might be suspicious of ECD. In January 2014, the patient was re-admitted to the hospital because multiple bone lesions were newly detected and cytopenia could still be detected in the peripheral blood examination. We performed bone marrow examination, bone biopsy, and lymph node biopsy for diagnosis of ECD. In addition, we reviewed the bone marrow results examined in March 2009 with additional immunohistochemical stain, and we were finally able to confirm the diagnosis of ECD. Thus, cytopenia resulted from bone marrow involvement of ECD. However, axial skeleton and lymph node involvement were detected 5 years after the initial presentation. Unlike other cases of ECD, our patient developed slow progression during a period of more than 5 years. We thought that the cause of this slow progression may be that despite having bone marrow involvement, there was no vital organ dysfunction.
ECD presenting with cytopenia resulting from bone marrow involvement, axial skeletal lesions rather than peripheral bone lesions, and involvement of lymph nodes have been rarely described in the literature. Thus, we report on a rare case of ECD with axial skeleton, lymph node, and bone marrow involvement.