Introduction
Soft tissue sarcomas (STS) form a set of heterogeneous neoplasms originating from mesenchymal cells. They are rare tumors comprising approximately 1% of all adult malignancies and 12% of pediatric cancers [1]. STS have different tumor biology, clinical behavior, and response to treatment, and some occur mainly in childhood, while others are unusual in young children [2]. Some STS, including rhabdomyosarcoma (RMS), Ewing sarcoma (EWS), primary neuroectodermal tumor (PNET), and desmoplastic small round cell tumor (DSRCT), are most common in children but occur rarely in adults. However, risk of mortality and morbidity is higher in adults with pediatric sarcoma with a comparable diagnosis [3]. Due to the rarity of adult STS, information regarding its clinical and biological features is limited; there is a lack of information on the survival of adults with STS that occur in the pediatric population.
RMS is the most frequent tumor in children, accounting for more than 50% of cases [4]. Among three types of RMS (alveolar, embryonal, and pleomorphic), pleomorphic RMS is most common in adults, and tends to occur in the lower extremity. The family of EWS includes EWS and PNET. The majority of patients with EWS and PNET are younger than 30 years of age; they are small round-cell tumors consisting of undifferentiated cellswith uniform nuclei and scanty cytoplasm [5]. For patients with localized disease, survival can be achieved for up to 70%; however, a relapse rate of up to 30% has also been reported [6]. DSRCT is a rare but highly aggressive mesenchymal tumor that develops in the abdominal cavity of young male adults [7]. It usually develops in adolescents and young adults (AYA), with a mean age at diagnosis of approximately 22 years. The prognosis is particularly poor, largely due to the fact that the majority of patients present with metastatic disease.
Previous studies on treatment outcome of pediatric-type sarcoma reported controversial results on age as a predictor of poor prognosis. A study comparing adult and pediatric RMS from 1973 to 2005 reported that adults had worse survival than children with similar tumors [3]. In pediatric trials, older age has shown an association with worse outcome, and unfavorable histology and distant metastasis were more common in adults [8]. However, the prognosis and optimal treatment strategies for the adult patient population are still not conclusive.
In this study, we will compare features of four types of pediatric sarcomas that occur in both adults and children, characterize clinical outcome, and identify the prognostic factors associated with survival.
Materials and Methods
1. Study population
The clinical and survival outcome data of patients with a reported diagnosis of RMS, EWS, PNET, and DSRCT between 1985 and 2011 were obtained from the database at Severance Hospital. All patients were treated with a multidisciplinary approach, including surgery, chemotherapy, or radiotherapy. Clinical data of these patients were reviewed retrospectively. The following clinical parameters were collected: demographic data, pathology, primary anatomic site, tumor extent at diagnosis, chemotherapy, radiotherapy, surgery, recurrence or progression, and survival. The following prognostic factors were analyzed: age, sex, tumor histology, primary site, tumor extent, primary tumor size, surgery, chemotherapy, and radiotherapy. Favorable tumor sites were defined as nonparameningeal head and neck, genitourinary sites except bladder and prostate, and orbit. Unfavorable sites included the parameningeal head and neck region, bladder, prostate, limb, and other sites. Tumor extent was defined using the Intergroup Rhabdomyosarcoma Study Group.
2. Chemotherapy
Most study patients (90.4%) received chemotherapy and the most common chemotherapy regimen was VP16/adriamycin/cyclophosphamide. Additional regimens included combinations of adriamycin, ifosphamide, vincristine, actinomycin-D, and cisplatin.
3. Radiotherapy
Radiotherapy (RT) techniques and methods evolved with technology over the course of the study; however, the basic principles were applied to all patients as follows. Patients’ extremities were immobilized for simulation and treatment in custom molds. The target volume encompassed the entire affected compartment, but was longitudinally extended 5 to 8 cm beyond the tumor. If necessary, additional magnetic resonance imaging was used for enhanced target definition. Most patients received a consistent dose/fractionation schedule of 50.4 Gy at 1.2 Gy fractions. The median RT dose was 45 Gy (range, 14 to 85 Gy).
4. Surgery
Debulking surgery, or wide excision, was performed in 129 patients (58.6%). Wide excision refers to a dissection plane through unaffected normal tissue within the involved compartment, as defined by the criteria of Enneking et al. [9].
5. Statistical analysis
Overall survival (OS) and event-free survival (EFS) were estimated using the Kaplan-Meier method. OS was calculated from the date of diagnosis to the date of death from the disease or the last follow-up. EFS was calculated from the date of treatment to the first documented relapse or progression. Survival estimates were calculated using the Kaplan-Meier method and log-rank test was used for comparison of survival curves. The chi-squared test was used for comparison of clinical parameters. Multivariate survival analyses using a Cox’s proportional hazard model were performed in order to characterize prognostic factors for OS and EFS.
Results
1. Clinical characteristics of pediatric sarcoma in children and adults
Results of comparison of the clinical findings in children and adult patients are shown in Table 1. A total of 220 patients with a reported diagnosis of RMS, EWS, PNET, and DSRCT were analyzed. The median age at the time of diagnosis was 15.6 years (range, 0 to 81 years). There were 108 pediatric patients (49%) and 112 adult patients (51%). There was a statistically significant difference in the sex of patients: there were more male than female patients in the adult population (p=0.010). No differences in the baseline characteristics, except sex, were observed between children and adults. More than half of the tumors in both children and adults were located at unfavorable sites (68.5% and 68.7%). A total of 158 patients (71.8%) presented with localized disease, 59 patients (26.8%) with metastatic disease and 3 patients (1.4%) with unknown status. Tumor size was known for 216 patients, with 108 patients (49.1%) having tumor size less than 5 cm, and 108 patients (49.1%) having tumor size equal to or greater than 5 cm. Patients with metastatic disease had a higher proportion of large tumors than patients without metastatic disease (54% vs. 36.1%, p < 0.001). A total of 129 patients (59%) underwent debulking surgery. Similar rates of radiotherapy and chemotherapy were observed among children and adults. According to histological classification, 106 (48.2%) had RMS, 60 (27.3%) had EWS, 50 (22.7%) had PNET, and 4 (1.8%) had DSRCT.
For 108 children, with a median age of 6 years (range, 0 to 6 years), 74 patients (68.5%) had tumors located in unfavorable sites, 49 patients (45.4%) had tumor size equal to or greater than 5 cm, and 26 patients (24.1%) had metastatic disease. Sixty-one patients (56.5%) had undergone debulking surgery, 55 patients (50.9%) had received radiotherapy, and 95 patients (87.9%) had received chemotherapy. By histopathology, 56 patients (51.8%) had RMS, 30 (27.8%) had EWS, 18 (16.7%) had PNET, and 4 (3.7%) had DSRCT.
For 112 adults, with a median age of 26 years (range, 16 to 81 years), 77 patients (68.7%) had tumors located in unfavorable sites, 59 patients (52.7%) had tumor size equal to or greater than 5 cm, and 33 patients (29.5%) had metastatic disease. Thirty-nine patients (34.8%) had undergone debulking surgery, 63 patients (56.3%) had received radiotherapy, and 100 patients (89.3%) had received chemotherapy. By histopathology, 50 patients (44.6%) had RMS, 30 (26.8%) had EWS, 32 (28.6%) had PNET, and none (0%) had DSRCT.
2. Follow-up and treatment outcome
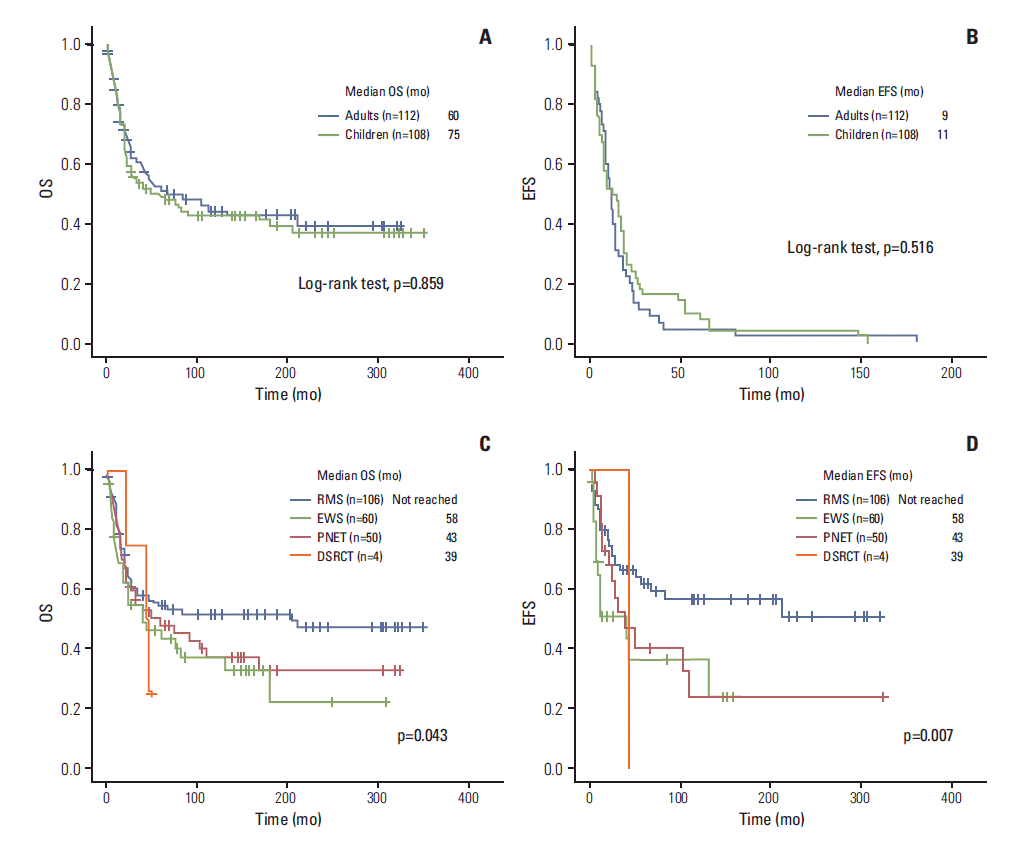
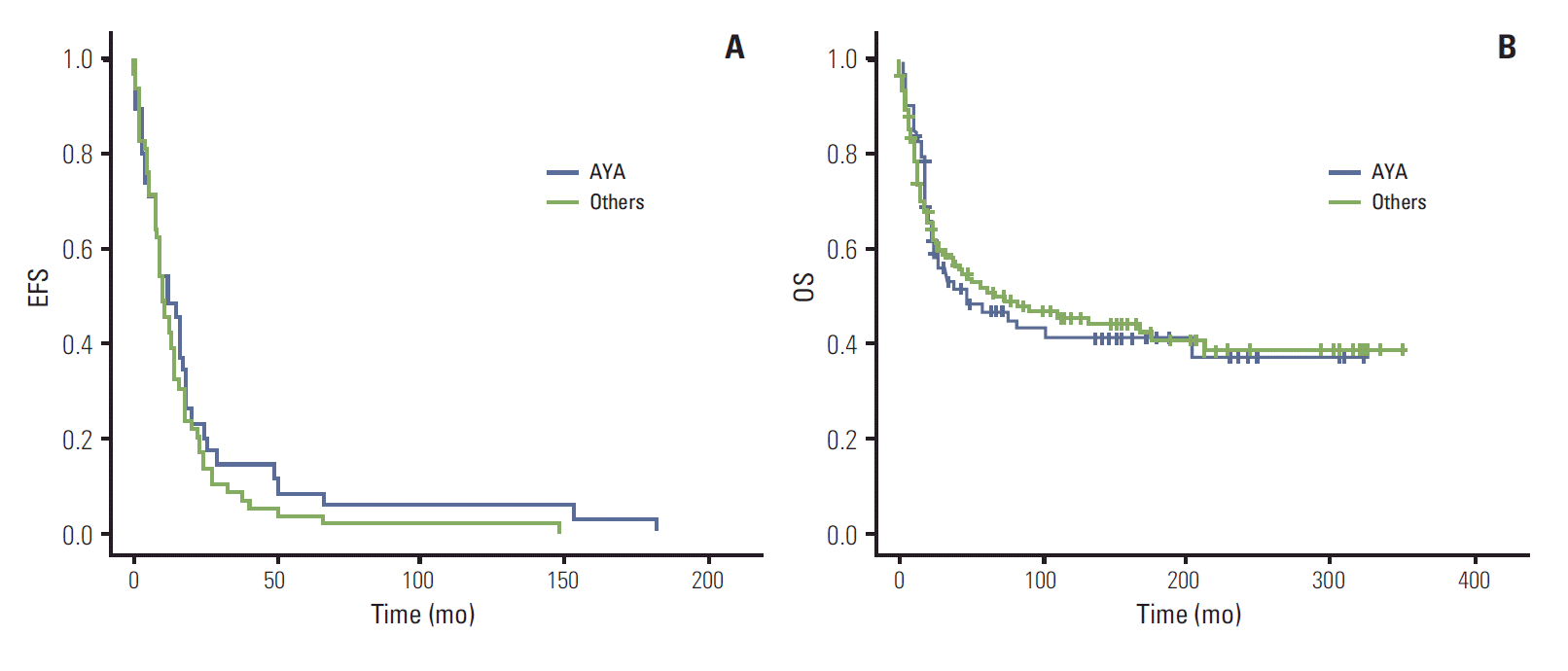
Among 220 patients, 115 (52.3%) had died at the time of analysis with a median follow-up period of 6.6 years (range, 0.1 to 29.0 years). Estimated median OS of all patients was 75 months (95% CI, 27.2 to 122.8 months), and the median EFS of all patients was 11 months (95% CI 8.8 to 13.2 months). No significant difference in OS was observed between adults and children (60 months [95% CI, 1.9 to 132.1] vs. 75 months [95% CI, 11.6 to 104.3], p=0.859) (Fig. 1A). In addition, no difference in EFS was observed between adults and children (9 months [95% CI, 9.4-14.6] vs. 11 months [95% CI, 5.1 to 18.9], p=0.516) (Fig. 1B). No significant difference in OS and EFS was observed among patients with localized disease only. The 5-year OS rates for RMS, PNET, EWS, and DSRCT were 45%, 30%, 33%, and 0%, respectively. In comparison of survival outcomes according to histology, a significant difference in median OS was observed among the four groups (not reached vs. 58 months [95% CI, 0 to 122.9] vs. 39 months [95% CI, 3.6 to 74.3] vs. 43 months [95% CI, 17.5 to 68.4], p=0.043) (Fig. 1C). In comparison of EFS among children, a significant difference was still observed among the four groups (not reached vs. 38 months [95% CI, 1.5 to 68.6] vs. 39 [95% CI, 6.4 to 89.6] vs. 43 [95% CI, 11.6 to 104.3], p=0.007) (Fig. 1D). No significant difference in OS was observed among adults according to histologic subtypes (not shown). We then evaluated survival outcome of AYA populations only. The EFS of the AYA population was 12 months (95% CI, 6.8 to 17.1 months) and that of the rest of the population was 10 months (95% CI, 7.5 to 12.5 months). The OS of the AYA population was 60 months (95% CI, 0 to 96.3 months) and that of the rest of the population was 77 months (95% CI, 6.4 to 147.6 months). No significant difference in both EFS and OS was observed between these two populations (Appendix 1A and B).
3. Analysis of prognostic factors
We performed univariate and multivariate analyses using the log-rank test for analysis of prognostic factors in adults and children (Table 2). In multivariate analysis, distant metastasis (HR, 1.617; 95% CI, 1.022 to 2.557; p=0.040) and no debulking surgery (HR, 1.443; 95% CI, 1.104 to 1.812; p=0.012) showed independent association with worse OS. In children, tumor size of more than 5cm (hazard ratio [HR], 1.540; 95% CI, 1.06 to 2.24; p=0.024), distant metastasis (HR, 1.851; 95% CI, 1.249 to 2.745; p=0.001), no debulking surgery (HR, 1.811; 95% CI, 1.467 to 1.958; p=0.006), and PNET histology (HR, 2.387; 95% CI, 1.230 to 4.631; p=0.010) showed significant association with worse OS. In addition, PNET showed an association with poor survival (HR, 2.387; 95% CI, 1.230 to 4.631; p=0.010). For adults, no debulking surgery (HR, 1.605; 95% CI, 1.349 to 1.910; p=0.003) was the only poor prognostic factor of OS. In multivariate analysis among adults, tumor size, distant metastasis, and histopathology did not predict poor outcomes.
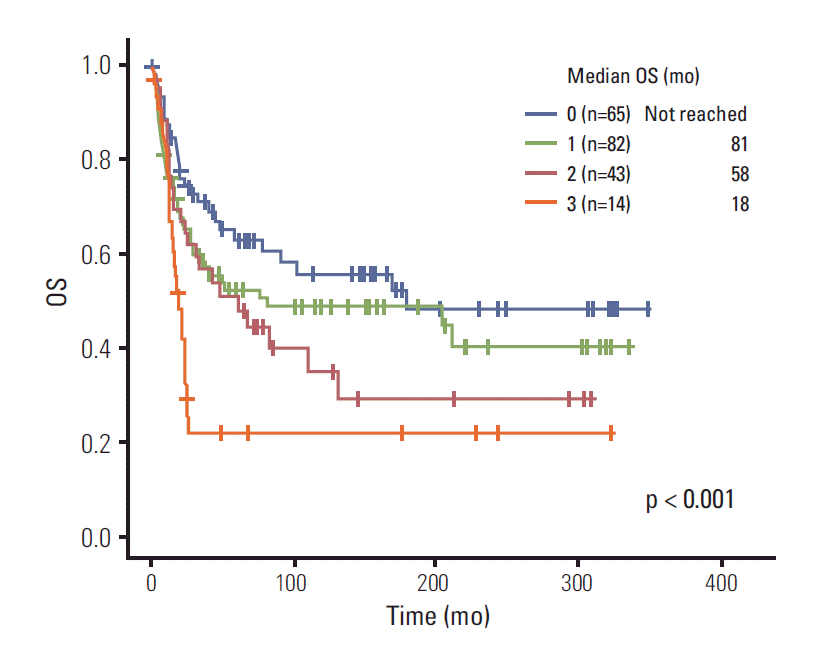
Next, we compared OS of patients according to number of poor prognostic factors. Poor prognostic factors were scored as follows: no distant metastasis=0, distant metastasis=1; tumor size less than 5 cm=0, tumor size equal or greater than 5 cm=1; surgery=0, no surgery=1. The Kaplan-Meier curve of four stratified scores showed a significant difference in survival (0 [not reached] vs. 1 [81 months] vs. 2 [58 months] vs. 3 [18 months], p < 0.001) (Fig. 2).
In a subgroup analysis of 158 patients with localized disease, 52 patients (32.9%) showed recurrence: 21 patients (13.3%) with local recurrence and 31 patients (19.6%) with distant recurrence. The most frequent sites of distant recurrence were lung (15%), brain (7%), bone (6%), spine (2%), and liver (1%), in the order of frequency.
Discussion
Using data during a 26-year period, we described 220 adult and pediatric patients with a reported diagnosis of RMS, EWS, PNET, and DSRCT. To the best of our knowledge, our work represents the first study examining the survival and prognostic factors in both pediatric and adult patients in the Asian population. While there were no significant differences in baseline characteristics in the study population, no significant differences in OS and EFS were observed between adults and children. In children, large tumor size (> 5 cm), metastatic disease, no surgical treatment, and PNET histology showed an association with shorter OS, whereas in adults, only no debulking surgery showed an association with shorter OS.
The effect of age on survival of sarcoma patients has been a subject of debate, with different conflicting results from different studies. In a few retrospective studies, survival in adults was reported to be dramatically worse than that reported for children [3,10,11]. Sultan et al. [3] reported that adults with RMS had worse survival than children with similar tumors. Lee et al. [12] reported that adults have fared worse than children due to less aggressive treatments, and fewer adults were treated with chemotherapy. However, in recent studies, age was not found to be a predictor of poor prognosis in adults [13,14]. In our study, we also did not observe a difference in survival outcome among children and adults. The differences in underlying tumor biology between adults and children are not yet known. It may be that the disease is more aggressive in adults, or that adults do not respond favorably to the current treatment regimens [15]. Conduct of further clinical and molecular studies is warranted in order to explain the possible differences between adults and children.
In the multivariate model, we compared prognostic factors of survival between adults and children. In all patients, surgical intervention was found to show significant association with improved survival when compared with no surgical intervention. Surgery is the mainstay of treatment of STS, and the rate of local recurrence following wide resection with negative margins is usually below 20% [16]. However, 37.3% of patients did not undergo surgery due to unresectability of the tumor and co-existing distant metastases. In univariate analysis, adjuvant RT did not show significant association with improved survival, consistent with previous data indicating that RT improves local control, but not OS [17]. Chemotherapy also did not show significant association with improved survival, however, there are issues to consider. Most patients received either adjuvant or palliative chemotherapy, and only 21 patients (9.5%) did not receive chemotherapy. It may be that there was not enough power to make a meaningful comparison between those who received chemotherapy and those who did not, or that patients who did not receive chemotherapy had less aggressive disease and better control with surgery alone. In addition, due to the retrospective nature of data collection, there was insufficient information on the chemotherapeutic regimen; therefore, it was difficult to estimate dose intensity of chemotherapy. The impact of histology on OS was only significant for PNET, which showed an association with poor survival outcome in children.
We reviewed previous studies of pediatric-type sarcoma in adults and children, as shown in Table 3 [3,18-24]. Most studies were confined to a single disease and 5-year survival rate ranged between 26% and 61%. Our study showed a 5-year survival rate of 37% with different survival outcomes according to histologic types. As shown in Table 3, previous studies have reported 5-year OS rates ranging between 20% and 40% in adult RMS. Comparable to these results, in our series, the 5-year OS rate of RMS adult patients was 45%.
Recently, increased understanding of the molecular biology of sarcomas has led to advances in molecular diagnostics and clinical management of certain sarcomas [25]. Molecular diagnostics have identified sarcomas with specific genetic alterations such as reciprocal translocations resulting in oncogenic fusion transcripts and specific oncogenic mutations. For example, the unique translocation found in DSRCT involves the EWSR1 and WT1 genes. The EWSR1-WT1 fusion protein acts as an oncogene and several transcriptional targets have been identified, such as platelet derived growth factor A and insulin-like growth factor 1 receptor. However, their precise contribution to transformation and their potential as a therapeutic target is unknown [7]. Conduct of further investigations exploring values of molecular markers will be necessary in order to improve survival outcome.
Our study has a few limitations. First, it is retrospective and the data collected often lacked detailed information on treatment. A lack of details of systemic chemotherapy and a few missing data are major limitations of our findings. Second, as it spanned a long period of time from 1985 until now, the heterogeneity in treatment strategies could result in different survival outcomes regardless of initial disease presentation. Third, due to the heterogeneity of histologic subtypes, it may be difficult to draw a common conclusion from the analysis of prognostic factors. Despite these limitations, our study is unique in that we have compared the survival outcome of a large number of adults and children with similar baseline characteristics.
Conclusion
Our study adds relevant data on clinical features and outcome of pediatric tumor in adult patients. No significant difference in survival outcome was observed between adults and children. Metastatic disease and no surgical treatment are poor prognostic factors for OS among pediatric-type sarcomas for both adults and children. Additional studies on treatment outcomes and molecular biology are clearly needed in order to further clarify differences between adult and pediatric sarcoma.