Transcription Factors in the Cellular Signaling Network as Prime Targets of Chemopreventive Phytochemicals
Article information
Abstract
Accumulating evidence from epidemiologic and laboratory studies support an inverse relationship between a regular consumption of fruits and vegetables and the risk of specific cancers. Numerous phytochemicals derived from edible plants have been reported to possess ability to interfere with a specific stage of carcinogenic process. Multiple mechanisms have been proposed to account for the anti-carcinogenic actions of dietary constituents, but more attention has recently focussed on intracellular signaling cascades as common molecular targets of a wide variety of chemopreventive phytochemicals.
Search for Chemopreventive Substances from Edible Plants
Cancer chemoprevention is an attempt to use either naturally occurring or synthetic substances or their mixtures to intervene in the progress of carcinogenesis, before the malignancy manifests. The term 'chemoprevention' was coined in the mid 1970s by Michael B. Sporn, who is an innovator in cancer prevention research. Numerous chemical substances have been found to prevent or halt carcinogenesis, and it is noticeable that a substantial body of chemopreventive agents are derived from our ordinary foods, particularly vegetables and fruits. Examples are garlic, soya, ginger, onion, broccoli, cabbage, cauliflower, Brussels sprouts, and turmeric.
A large number of population-based studies have highlighted the ability of macronutrients and micronutrients contained in vegetables and fruits to reduce the risk of cancer. The most exciting findings have been achieved with antioxidant vitamins and their precursors, which are abundant in dark, leafy green vegetables and yellow orange fruits and vegetables. However, plants contain numerous chemical substances other than antioxidant vitamins or minerals that might also be useful in preventing cancer. Recently, the focus and emphasis have shifted to these non-nutritive components in the plant-based diet, collectively termed phytochemicals ('phyto,'- from the Greek word meaning plant), which possess substantial anti-carcinogenic and anti-mutagenic properties (1). Currently a series of human intervention trials are under way with nutritional supplements and modified diets to prevent cancer, and it is expected that someday people will take specially formulated pills containing individual chemopreventive phytochemicals or their mixtures for the purpose of avoiding cancer or delaying at least its onset. However, precise assessment of underlying mechanisms of individual ingredients is necessary before they can be recommended for inclusion in dietary supplements or tested in human intervention trials.
Given the great structural diversity of phytochemicals, it is not feasible to define structure-activity relationships to deduce their underlying molecular mechanisms. A better approach to studying the mechanisms of chemopreventive phytochemicals is to assess their effects on the deregulated cellular signal transduction pathways leading to precancerous or cancerous status (1,2). The scope of this review is limited to effects of representative chemopreventive phytochemicals on activation or induction of transcription factors with particular emphasis given to NF-κB, AP-1 and Nrf2.
Biochemical Basis of Chemoprevention with Edible Phytochemicals
Carcinogenesis has been recognized as a gradual, stepwise process, rather than a single event. From the study of experimentally induced carcinogenesis in rodents, tumor development is considered to consist of several distinct, but closely linked stages-initiation, promotion and progression. Initiation is a rapid and irreversible process that involves a chain of extracellular and intracellular events. These include the initial uptake of or exposure to a carcinogenic agent, its distribution and transport to organs and tissues where metabolic activation and detoxification may occur, and the covalent interaction of reactive species with target cell DNA, leading to genotoxic damage. In contrast to initiation, promotion is considered to be a relatively lengthy and reversible process in which actively proliferating preneoplastic cells accumulate. Progression, the final stage of neoplastic transformation, involves the growth of a tumor with invasive and metastatic potential. Although such divisions may be an oversimplification of carcinogenesis, the multi-stage nature of carcinogenesis offers ample opportunities for intervention-a chance to prevent, slow or even halt the gradual march of normal, healthy cells towards malignancy.
According to the conventional classification originally proposed by Lee Wattenberg, chemopreventive agents are subdivided into two main categories, i.e., blocking agents and suppressing agents (3). Blocking agents prevent carcinogens from reaching the target sites, from undergoing metabolic activation or subsequently interacting with crucial cellular macromolecules, such as DNA, RNA and proteins (Fig. 1). Suppressing agents, on the other hand, inhibit the malignant transformation of initiated cells, in either the promotion or the progression stage. Chemopreventive phytochemicals can block or reverse the early precancerous stages (initiation and promotion). They can also halt or at least retard the development and progression of precancerous cells into malignant ones with increased invasiveness and metastatic potential (Fig. 1). The effects of any single chemopreventive phytochemical on tumor development, however, are usually considered to be the outcome of the combination of numerous effects, rather than a single biological response. Therefore, it is important to identify molecular targets that are associated with each stage in the natural history of cancer and are modulated by chemopreventive phytochemicals. Table 1 illustrates the chemical source of some of the most extensively investigated chemopreventive phytochemicals and their dietary sources. The cellular and molecular events modulated by these dietary chemopreventive phytochemicals include carcinogen activation/detoxification by xenobiotic-metabolizing enzymes; DNA repair; cell-cycle progression; cell proliferation and differentiation; apoptosis; expression and functional activation of oncogenes and tumor-suppressor genes; angiogenesis and metastasis; hormonal and growth factor activity, etc.

Dietary phytochemicals blocking or suppressing multi-stage carciniogenesis. Certain chemopreventive phytochemicals in the diet inhibit metabolic activation of the procarcinogens to ultimate electrophilic species and/or their covalent interaction with target cell DNA, thereby blocking the initiation (blocking agents). Alternatively dietary blocking agents can stimulate the detoxification of the pro- or ultimate carcinogens. Others suppress the later steps (promotion and progression) of multi-stage carcinogenesis (suppressing agents). Some phytochemicals can act as both blocking and suppressing agents. Adapted from reference 1.

Edible phytochemicals with chemopreventive potential
Cellular Signaling Molecules as Targets of Chemopreventive Phytochemicals
Remarkable progress in cellular and molecular biology over the past three decades led to have a deep insight into the biochemical events associated with the multistage process of carcinogenesis. Now we are better aware of how certain dietary phytochemicals are able to alter this process (Fig. 1). Despite such progress, the identification of molecular and cellular targets of chemopreventive phytochemicals is still incomplete. Majority of the molecular alterations linked to carcinogenesis occur in intracellular signal transduction pathways responsible for regulating proliferation and differentiation of target cells. One of the central components of the cell signaling network that maintains homeostasis is the family of proline-directed serine/threonine kinases named the mitogen-activated protein kinases (MAPKs).
Abnormal or improper activation or silencing of the MAPK pathway or its downstream transcription factors can result in uncontrolled cell growth, leading to malignant transformation. Some phytochemicals 'switch on' or 'turn off' the specific signaling molecule(s) depending upon the nature of the cascade they target, preventing abnormal cell proliferation and growth (4~12). Cell signaling kinases other than MAPKs, such as protein kinase C (PKC) and phosphoinositide-3-kinase (PI3-K), are also important targets of certain chemopreventive phytochemicals. These upstream kinases activate a distinct set of transcription factors including nuclear factor kappaB (NF-κB) and activator protein 1 (AP-1).
NF-κB and AP-1
Numerous intracellular signal transduction pathways converge with the activation of the transcription factors NF-κB and AP-1, which act coordinately to regulate target gene expression (Fig. 2). Aberrant activation of NF-κB has been associated with protection against apoptosis and stimulation of proliferation in malignant cells (13,14). Further studies have shown that overexpression of NF-κB is causally linked to the phenotypic changes characteristic of neoplastic transformation (15). A relatively large number of chemopreventive phytochemicals derived from diet have been shown to suppress constitutive NF-κB activation in malignant cells or induced by the external tumor promoter phorbol 12-myristate 13-acetate (PMA) or tumor necrosis factor-α (TNF-α) (11,16,17).

The schematic representation of signalling cascades and activation of NF-κB and AP-1. The activation of NF-κB and AP-1 begin, in general, with stimulation of specific receptors at the cell surface and recruitment of adaptor proteins, which targets the external signal for specific transduction pathways controlled by various kinases. The NF-κB pathway converges upon a high-molecular mass multiprotein complex named IκB kinase (IKK) signalsome, which, in turn, promotes the phosphorylation of IκB. This targets IκB for ubiquitination and subsequent degradation by 26S proteasome. As a consequence, NF-κB is released and rapidly translocated to the nucleus where it binds to specific promoter regions of various genes. The IKK signalsome is activated by the NF-κB inducing kinase (NIK), which lies immediately upstream of IKKs in the NF-κB activation cascades. NIK mutants block TNF-α-and IL-1-induced NF-κB activation. Although the upstream signalling pathways that regulate NIK has not been clarified yet, there is some evidence to support the involvement of MAPK/ERK kinase kinase-1 (MEKK1), a kinase upstream of ERK as well as p38 MAPK. Many studies have revealed a close association between the MAPK activity and phosphorylation and degradation of IκB protein, which facilitates nuclear translocation and subsequent DNA binding of NF-κB in various cell systems. Recent reports showed that the NF-κB activation is also regulated by the Akt signaling pathway (46,47). Phosphatidylinositol 3-kinase (PI3-K) activates Akt/protein kinase B via phosphorylation by 3-phosphoinositide-dependent protein kinase-1 (PDK1). Genistein specifically inhibits Akt activity and Akt-mediated NF-κB activation. Epiegallocatechin gallate (EGCG) can block the activities of PI3-K and Akt. There is crosstalk between the Akt and NF-κB signaling pathways- Akt phosphorylation leads to activation of NF-κB by stimulating IκB kinase (IKK) activity. IKK is also a target of chemopreventive phytochemicals, including curcumin, resveratrol and EGCG (see text for details). AP-1 heterodimers are constitutively localized within the nucleus, and transactivation of AP-1 is achieved through phosphorylation of its activation domain by MAPK. The MAPK family proteins have been shown to play a key role in regulating AP-1 activation in various types of cells in culture. AP-1 represents a heterogenous set of dimeric proteins consisting of members of Jun, Fos, and ATF families. AP-1 mRNA expression is dependent on MAPKs: ERK1/2 phosphorylates Elk-1, JNK phosphorylates ATF-2 and c-Jun in c-fos and c-jun induction, respectively, while p38 phosphorylates both Elk-1 and ATF-2. c-Fos and c-Jun form heterodimers and bind to AP-1 response element in the promoter of target genes. The target molecule/event blocked/inactivated by chemopreventive phytochemicals is marked with a symbol (⦸).
AP-1 is another transcription factor that regulates expression of genes that are involved in cellular adaptation, differentiation and proliferation. Functional activation of AP-1 is considered to be an important event in signal transduction associated with malignant transformation as well as tumor promotion (18~21). AP-1 consists of either homo- or heterodimers between members of the Jun and Fos families, which interact via a leucinezipper domain. This transcription factor is also regulated by the MAPK signaling cascade (21~23).
As NF-κB and AP-1 are ubiquitous eukaryotic transcription factors that mediate pleiotropic effects of both external and internal stimuli in the cellular signalling cascades, they are prime targets of diverse classes of chemopreventive phytochemicals.
Chemopreventive Phytochemicals Targeting NF-κB and AP-1
1) Curcumin
Curcumin, a yellow pigment present in the rhizome of turmeric (Curcuma longa L., Zingiberaceae) and related species, is one of the most extensively investigated phytochemicals, with regard to chemopreventive potential. One plausible mechanism underlying chemopreventive properties of curcumin involves suppression of tumor promotion, particularly in mouse skin carcinogenesis. Pretreatment of human colonic epithelial cells with curcumin resulted in marked inhibition of TNF-α-induced cox-2 gene transcription and NF-κB activation (24). In this study, curcumin inhibited IκB degradation by downregulation of NIK and IKKα/β.
When curcumin was applied topically to dorsal skin of female ICR mice, it prevented the PMA-induced activation of both NF-κB and AP-1 (25). The inhibition was accompanied by blockade of degradation via phosphorylation of IκBα and also by reduced nuclear translocation of the p65 subunit of NF-κB (26). Topically applied curcumin caused inhibition of catalytic activity of epidermal ERK1/2, which may account for its inactivation of NF-κB and COX-2 (26).
Curcumin suppressed the PMA-induced nuclear translocation and DNA binding of NF-κB in human myeloid leukemia cell line by blocking phosphorylation and subsequent degradation of IκB (27). PMA- and hydrogen peroxide-induced activation of NF-κB was similarly attenuated by curcumin treatment. In addition, curcumin inhibited IκBα phosphorylation in malignant cells (28,29) through suppression of IKK activity, which contributed to its antiproliferative, proapoptotic and antimetastatic activities.
2) Epigallocatechin gallate (EGCG)
EGCG is an antioxidant and chemopreventive polyphenol that is found in green tea. It has been shown to suppress malignant transformation in PMA-stimulated mouse epidermal JB6 cell line, which appeared to be mediated by blocking AP-1 (30) and NF-κB (31) activation. More recently, EGCG treatment of human epidermal keratinocytes resulted in significant inhibition of UVB-induced activation of IKKα, phosphorylation and subsequent degradation of IκBα and nuclear translocation of p65 (32). In the H-ras-transformed epidermal JB6 cells, EGCG, inhibited ras-activated AP-1 activity (33,34). Similar AP-1 inhibition was observed in the epidermis of transgenic mice that harbour an AP-1-driven luciferase reporter gene. A recent studies from this laboratory has demonstrated the inhibition by EGCG of COX-2 expression in PMA-stimulated mouse skin and human mammary epithelial MCF-10A cells (35). Suppression of PMA-induced COX-expression by EGCG appears to be associated with its inhibition of DNA binding and transcriptional activity of NF-κB in MCF-10A cells (H.-K. Na and Y.-J. Surh, unpublished data).
Nomura and colleagues (36) have reported the inhibitory effect of EGCG on UV-induced PI3-K activation in mouse epidermal cells. The reduction of signalling via the PI3-K, then AKT and finally to the NF-κB pathway by EGCG was reported to be mediated through inhibition of Her-2/neu receptor tyrosine phosphorylation (37). EGCG also inhibited vascular endothelial growth factor (VEGF) production by inhibiting both constitutive activation of Stat 3 and NF-κB, but not ERK or Akt in human breast and head and neck cancer cell lines (38).
EGCG treatment resulted in inhibition of cell growth, G0/G1-phase arrest of the cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells, but not in normal human epidermal keratinocytes (39). A431 cells were more prone to EGCG-induced suppression of constitutive NF-κB activation compared with normal human epidermal keratinocytes, indicating that EGCG-mediated cell cycle deregulation and apoptosis of cells might be attributable to its inactivation of NF-κB. The roles of EGCG and other tea polyphenols on cellular signaling have been recently reviewed (40,41).
3) Genistein
Genistein, a soy-derived isoflavone, is considered to contribute to the putative breast and prostate cancer preventive activity of soya. Genistein inhibited PMA-induced AP-1 activity, expression of c-Fos, and ERK activity in certain human mammary cell lines (42). Genistein treatment abrogated not only NF-κB DNA binding, but also activation of Met receptor signaling in human hepatocarcinoma HepG2 cells stimulated with hepatocyte growth factor (43). The downregulation of c-jun and c-fos by genistein was also observed in UV-stimulated SENCAR mouse skin (44).
Genistein at the apoptogenic concentration also inhibited the H2O2- or TNF-α-induced activation of NF-κB in both the androgen-sensitive (LNCaP) and -insensitive (PC3) human prostate cancer cell lines by reducing phosphorylation of IκBα and the nuclear translocation of NF-κB (45). Genistein-mediated inactivation of NF-κB was associated with specific inhibition of Akt activity and abrogation of EGF-induced activation of Akt in the prostate cancer cells (46) and mammary cancer (47). The same studies also revealed that Akt transfection led to the activation of NF-kB which was completely blocked by genistein treatment, suggesting that inhibition of the cross-talk between Akt and NF-κB could provide a novel mechanism responsible for proapoptotic activity of genistein, preferentially towards tumorigenic but not normal prostate epithelial cells.
PMA- or TNF-α-induced NF-κB DNA binding and NF-κ B-derived COX-2 promoter activity as well as COX-2 expression were inhibited in human alveolar epithelial carcinoma cells by genistein treatment (48). In human U937 monocytes, genistein exerted no substantial inhibitory effect on DNA binding of NF-κB yet markedly attenuated its transcriptional activity (49). Consistent with this notion, preliminary data from this laboratory demonstrate that genistein strongly suppress NF-κB transcriptional activity in PMA-stimulated human mammary epithelial cells as determined by the luciferase reporter gene assay, but failed to interfere with IκB degradation and nuclear translocation and DNA binding of NF-κB (M.-H. Chung and Y.-J. Surh, manuscript in preparation). Genistein, without influencing the IKK activity, can block the phosphorylation of p65 subunit of NF-κB, thereby hampering its interaction with coactivators such as cyclic AMP-response element binding protein-binding protein (CBP/p300), a key element of the transcription initiation complex that bridges DNA bound transcription factors to the transcription machinery as illustrated in Fig. 3. AP-1 transcriptional activity might be similarly down-regulated by genistein and other phytochemicals that interfere with AP-1 binding to CBP/p300.

Proposed pathways for CBP- and kinase-dependent transcriptional activation of NF-κB and AP-1. A number of extracellular stimuli triggered activation of specific kinases that are involved in transcriptional activation of NF-κB and AP-1. IKK phosphorylates IκB, which results in degradation of IκB by the proteasomes, releasing p65, which then translocates to the nucleus. Some kinases phosphorylate p65, the functionally active NF-κB transactivation subunit. Phosphorylated p65 enhances NF-κB-dependent transcription, probably by influencing the binding affinities of p65 to coactivators such as p300/CBP (CREB-binding protein) or transcriptional initiation complex. CBP is a general coactivator protein that bridges DNA-bound transcription factors to the basal transcription machinery. In addition, CBP, having intrinsic acetyltransferase activity, acetylates p50 (NF-κBκDNA binding subunit). This causes increased affinity of NF-κB to DNA. On the other hand, JNK-induced phosphorylation of c-Jun, a component of the AP-1 complex, facilitates its interaction with p300/CBP. Abbreviations: GSK-3β,..glycogen synthase kinase-3; IκB, Inhibitor of NF-κB; MSK1, mitogen- and stress-activated protein kinase 1; P, phosphate; PKAc, catalytic subunit of protein kinase A; PKB/AKT, protein kinase B; JNK, c-Jun-N-terminal kinase; AP-1, activator protein-1.
4) Resveratrol
Resveratrol (3,4',5-trihydroxy-trans-stilbene) is a phytoalexin present in grapes (Vintis vinifera, Vitaceae) and a major antioxidant ingredient of red wine, it is responsible for so-called 'French paradox' Resveratrol treatment caused PMA-induced COX-2 expression as well as catalytic activity via cyclic AMP response element (CRE) in human mammary epithelial cells (50,51). It also inhibited PKC activation, AP-1 transcriptional activity and the induction of COX-2 promoter activity in PMA-treated cells. Resveratrol induced apoptosis while reducing the constitutive activation of NF-κB in both rat and human pancreatic carcinoma cell lines (52). Mammary tumors from animals treated with resveratrol displayed reduced expression of COX-2 and MMP-9 and NF-κB activation, compared with those from DMBA-treated controls (53). Treatment of human breast cancer MCF-7 cells with resveratrol also suppressed NF-κB activation and proliferation.
Treatment of androgen-sensitive prostate cancer cells (LNCaP) with resveratrol caused downregulation of prostate-specific antigen and p65, which was associated with activation of p53, p21WAF/CIP1 p300/CBP and Apaf-1 (54). Resveratrol-induced apoptosis in mouse JB6 epidermal cells was associated with phosphorylation of p53 which appeared to be mediated through activation of ERK and p38 (55). Yu and colleagues have demonstrated that resveratrol pretreatment gave rise to suppression of PMA- and UV-induced activation of AP-1 and MAPKs (ERK2, JNK1, and p38) in cultured HeLa, which was associated with inhibition of PKC and protein tyrosine kinase (56). Similarly, resveratrol blocked UV-induced activation of NF-κB through suppression of IKK activation (57). Resveratrol suppressed TNF-α-induced phosphorylation and nuclear translocation of p65, and NF-κB-dependent reporter gene transcription in myeloid leukemia cells (58). The suppression of NF-κB coincided with AP-1 inactivation. TNF-induced activation of MAPK kinase (MEK) and JNK was also abrogated by resveratrol (58). Resveratrol induced apoptosis in fibroblasts after the expression of H-ras, possibly through inhibition of NF-κB activation by blocking IKK activity (59).
Nrf2-Keap1 Signaling
A wide spectrum of xenobiotic metabolizing enzymes catalyze both phase I (oxidation and reduction) and phase II biotransformation (conjugation) reactions involved in carcinogen activation and/or deactivation. The phase II enzyme induction system represents an important component of the cellular stress response whereby a wide array of electrophilic and oxidative toxicants can be removed expeditionally prior to attacking the target cell DNA. Some chemopreventive phytochemicals with antioxidant activity exert their protective effects not only by scavenging reactive oxygen species (ROS), but also through induction of de novo expression of a battery of genes that encode detoxifying/defensive proteins including phase II enzymes. Several antioxidant-response element (ARE)-regulated gene products, such as glutathione S-transferase (GST), NAD (P)H:quinone oxidoreductase 1 (NQO), UDP-glucuronosyltransferase, γ-glutamate-cysteine ligase (GCL), and heme oxygenase-1 (HO-1), are known to mediate detoxification and/or to exert antioxidant functions, thereby protecting cells from genotoxic damage. The transcription of ARE-driven genes are regulated, at least in part, by nuclear transcription factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2), which is sequestered in cytoplasm by Kelch-like ECH-associated protein 1 (Keap1). Exposure of cells to ARE inducers results in the dissociation of Nrf2 from Keap1 and facilitates translocation of Nrf2 to the nucleus, where it heterodimerizes with small Maf protein, and bind to ARE, eventually resulting in the transcriptional activation of Nrf2-regulated genes (Fig. 4). The Nrf2-Keap1-ARE signaling pathway can be modulated by several upstream kinases including phosphatidylinositol 3-kinase, protein kinase C, and mitogen-activated protein kinases. Selected Nrf2-Keap1-ARE activators, such as oltipraz, anethole dithiolethione, sulforaphane, 6-methylsulphinylhexyl isothiocyanate, curcumin, caffeic acid phenethyl ester, 4'-bromoflavone, etc., are potential chemopreventive agents. Therefore, much attention has recently been focused on a chemopreventive strategy directed towards protection of DNA and other critical cellular molecules by inducing de novo synthesis of phase II detoxifying or antioxidant genes via the Nrf2-ARE core signaling pathway (60).
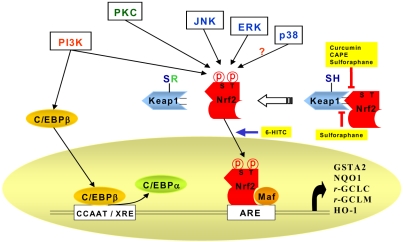
Signaling pathways involved in the Antioxidant responsive element (ARE)-mediated transcriptional response through the activation of Nrf2. A protein designated as Kelch-like-ECH-associated protein 1 (Keap1) has been shown to be a cytoplasmic repressor of Nrf2 and thus inhibits its ability to translocate to nucleus and transactivate the ARE. These two proteins interact with each other through the double glycine-rich domains of Keap1 and a hydrophilic region in the Neh2 domain of Nrf2. Keap1 contains many cysteine residues. Oxidants or phase II enzyme inducers can cause oxidation or covalent modification of these cysteine residues. As a result, Nrf2 is released from the repressor (Keap1). Dissociation of the Keap1-Nrf2 complex is also assumed to be facilitated through the phosphorylation of Nrf2 by upstream kinase(s) such as MAPK, PKC, or PI3-K. PI3-K is also considered to phosphorylate CCAAT/enhancer binding protein-β (C/EBP-β), inducing its translocation to the nucleus and binding to the CCAAT sequence of C/EBP-β response element within the xenobiotic response element (XRE), in conjunction with Nrf2 binding to ARE. Active C/EBPβ may compete with C/EBP-α for the C/EBP binding site. After nuclear translocation, Nrf2 associates with a small Maf protein, forming a heterodimer that binds to ARE, stimulating the ARE-driven expression of gene that encode phase-II detoxifying or antioxidant enzymes, such as glutathione S-transferase alpha2 (GSTA2), NAD(P)H:quinone oxidoreductase (NQO1), γ-glutamate cysteine ligase (γ -GCLC and γ -GCLM), and heme oxygenase-1 (HO-1). Nrf2 can also dimerize with c-Jun, ATF-4, PMF and PPAR-gamma, but the physiologic significance of Nrf2 dimerization with such different bZIP proteins remains to be clarified. Curcumin and caffeic acid phenethyl ester (CAPE) disrupt the Nrf2-Keap1 complex, leading to increased Nrf2 binding to ARE. Sulforaphane directly interacts with Keap1 by covalent binding to its thiol groups. 6-(Methylsulfinyl)hexyl isothiocyanate (6-HITC) - a sulforaphane analog derived from Japanese horseradish (wasabi)-stimulates nuclear translocation of Nrf2.
1) Nrf
A role for Nrf2 in the regulation of ARE-mediated gene expression has been demonstrated further in studies involving Nrf2-null mice (61). These mice failed to induce many of genes involved in carcinogen detoxification and protection against oxidative stress (61~66). Most notably, the Nrf2-null mice developed a larger number of tumors in the forestomach after treatment with the ubiquitous carcinogen benzo[a]pyrene, which was not prevented by oltipraz, a chemopreventive dithiolethione with phase II enzyme inducing activity (63,67). Nrf2-null mice were also have defects in detoxification of aflatoxin B1 (68). Stable transfection of L929 cells with a dominant negative mutant form of Nrf2 abolished induction of HO-1 by several toxicants (69). Fibroblasts from Nrf2(-/-) mice were found to express only about 15% of GCS mRNA compared to wild-type cells (70). Overexpression of Nrf2 led to activation of ARE-mediated transcription in human hepatoma (HepG2) cells (71).
2) Keap1 as a negative regulator of Nrf
A cytosolic actin-binding protein 'Kelch-like ECH-associated protein 1 (Keap1)' has been identified as a docking site where the bZIP proteins are sequestered under normal physiological conditions. Keap1 suppresses Nrf2 transcriptional activity by retaining the transcription factor in the cytoplasm, thereby hampering its nuclear translocation (Fig. 4).
The mechanisms by which cells recognize chemopreventive antioxidants or phase II enzyme inducers have not been fully elucidated. The Keap1-Nrf2 complex is an intracellular sensor for recognizing chemopreventive blocking agents or redox signalling from electrophiles or ROS (72). Many phase II gene inducers are generally electrophilic per se or can be readily convereted-nonenzymatically, via redox cycling- or metabolized to electrophilic intermediates in the body. Phase II enzyme inducers mimic prooxidants and electrophile although most of them are antioxidants by nature. Therefore, it might be more appropriate to call ARE as electrophile response element (EpRE)'. It is plausible that these reactive species interact with thiol groups of Keap1 and oxidize or covalently modify the cysteine residues within Keap1 and, possibly also Nrf2 (73~75), which would facilitate the release of Nrf2 from Keap1.
In accordance with this supposition, sulfhydryl reactive agents such as diethyl maleate abrogated Keap1 repression of Nrf2 activity, releasing the transcription factor (72). In this context, the cysteine residues in Keap1 may serve as a molecular sensor for the intracellular redox status, ensuring the proper and timely expression of genes involved in cellular antioxidant defense or detoxification of electrophilic toxicants.
3) Upstream signaling pathways regulating Nrf2 activation
Besides direct oxidation or covalent modification of the thiol groups of Keap1, the Nrf2-Keap1-ARE signaling can be modulated by post-transcriptional modification of Nrf2 as depicted in Fig. 4. Phosphorylation of Nrf2 at serine (S) and threonine (T) residues by kinases such as PI3-K, PKC, c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated protein kinase (ERK) is assumed to facilitate the release of Nrf2 from Keap1 and subsequent translocation by phosphorylation. p38 MAPK can either stimulate or inhibit the Nrf2 nuclear translocation depending on the cell type (76,77).
PI3-K also appears to play a role in Nrf2 activation in cells exposed to tert-butylhydroquinone (tBHQ), peroxinitrite, or hemin (78-81). In response to oxidative stress, the activation of signaling cascades mediated through PI3-K results in depolymerization of actin microfilaments thereby facilitating Nrf2 translocation to the nucleus (78). In addition, PI3-K also phosphorylates the CCAAT/enhancer binding protein-β (C/EBPβ), which then translocates to the nucleus and subsequently binds to the CCAAT sequence of C/EBP-β response element located within the xenobiotic response element (XRE), in parallel with Nrf2 binding with ARE (82,83). Transfection of human neuroblastoma cells with PI3-K activated ARE, which was attenuated by a pharmacological inhibitor of PI3-K or a dominant-negative mutant form of Nrf2 (79).
MAPKs, such as ERK, JNK, and p38 MAPK, are also considered to be involved in the ARE activation (84). Yu and colleagues reported that activation of MAPK signaling in Hep G2 cells induced ARE-mediated gene expression via a Nrf2-dependent mechanism (85), whereas p38 MAPK played an opposite role (76). However, Barogun and colleagues have addressed that the p38, but neither ERK nor JNK, is implicated in curcumin-mediated HO-1 gene induction via Nrf2-ARE in the rat kidney epithelial (NRK-52E) cells (77).
In addition, it has been reported that some other factors including p160 family of coactivators and CBP/p300 may interact with the Nrf2-Maf-ARE complex thereby enhancing transactivation of Nrf2 (86,87). A recent study shows that ERK and JNK signaling pathways induce the recruitment of co-activator to the transcription initiation complex and upregulate Nrf2 transcriptional activity (88).
Chemopreventive Phytochemicals Targeting the Nrf Signaling
Accumulating evidence supports that Nrf2-mediated activation of ARE, or more recently electrophile responsive element (EpRE), is a central part of molecular mechanisms governing the protective function of phase II detoxification and antioxidant enzymes against chemical carcinogenesis and oxidative stress. A wide variety of chemopreventive dietary phytochemicals that function as potent inducers of ARE-regulated gene expression have been shown to exert chemopreventive activities. Examples are sulforaphane, 6-methylsulphinylhexyl isothiocyanate (6-HITC), and curcumin, caffeic acid phenethyl ester (CAPE).
1) Curcumin
Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione], the yellow coloring pigment isolated from the rhizomes of the plant turmeric (Curcuma longa Linn, Zingiberaceae), has been shown to inhibit tumorigenesis in both initiation and promotion stages in several experimental animal models (89). Topical application of curcumin inhibited 7,12-dimethylbenz[a]anthracene (DMBA)-initiated and PMA-promoted skin tumorigenesis (90,91). In addition, dietary administration of curcumin protected against experimental carcinogenesis of other organs like forestomach, duodenum, and colon (92~94).
2) Caffeic acid phenethyl ester (CAPE)
CAPE, a phenolic constituent of the honeybee propolis, has anticancer and anti-inflammatory properties (95). It inhibits the development of azoxymethane-induced aberrant crypts in the colon of rats (96), and blocks tumorigenesis in a two-stage model of mouse skin tumorigenesis promoted by TPA (97).
Curcumin and CAPE disrupt the Nrf2-Keap1 complex formation, leading to increased Nrf2 binding to ARE (77, 98). In porcine renal epithelial cells, both phytochemicals elevated nuclear levels of Nrf2 by inactivating the Nrf2-Keap1 complex, which was associated with a significant increase in the activity and expression of HO-1 (77). p38 MAPK, which is upstream of Nrf2, seems to be involved in curcumin-induced HO-1 gene induction. In another study, curcumin increased the nuclear translocation of Nrf2, ARE DNA binding activity and GCL expression (98). It is notable that both curcumin and CAPE bear an α,β-unsaturated ketone moiety, and can therefore act as Michael-reaction acceptors capable of modifying cysteine thiols present in Keap1.
3) Sulforaphane and its analogues
Sulforaphane [1-isothiocyanato-(4R,S)-(methylsulfinyl) butane], a major isothiocyanate present in broccoli sprouts and mature broccoli, has been reported as a potent inducer of ARE-regulated enzymes (99~101). Sulphoraphane as well as phenethyl isothiocyanate differentially regulated the activation of MAPKs and Nrf2, ARE-mediated luciferase reporter-gene activity, and phase II enzyme gene induction (84,102). Sulphoraphane also directly interacts with Keap1 by covalent binding to thiol groups of this inhibitory protein (74). Analysis of gene-expression profiles by an oligonucleotide microarray revealed that sulforaphane upregulated the expression of NQO1, GST and GCL in the small intestine of wild-type mice, whereas the Nrf2-null mice displayed much lower levels of these enzymes (103). Sulforaphane has been shown to be protective against carcinogen-induced tumorigenesis in various experimental models (104~106). During extensive screening of vegetable extracts for GST-inducing activity in cultured rat liver epithelial RL-34 cells, Morimitsu and colleagues have identified a sulphoraphane analogue, 6-(methylsulphinyl)hexyl isothiocyanate (6-HITC) derived from Japanese horseradish wasabi (Wasabia japonica or Eutrema wasabi Maxim), as a key GST-inducer (107). 6-HITC stimulated nuclear translocation of Nrf2, which subsequently activated ARE. The compound also markedly induced both class α-GSTA1 and µ-GSTP1 isozymes in rat liver epithelial RL-34 cells via the Nrf2-ARE signal pathway. Oral administration of 6-HITC resulted in the induction of hepatic phase II detoxifying enzymes to a greater extent than sulphoraphane, whereas this induction was abrogated in Nrf2-null mice (107). Similarly, the chemopreventive efficacy of 6-HITC and other phase II enzyme inducers, such as oltipraz and sulphoraphane, was abolished in Nrf2-deficient mice (104,108), suggesting that Nrf2 is a critical molecular target for chemoprevention.
Future Perspectives
Over the past two and three decades, there has been a increasing number of chemopreventive phytochemicals identified in our daily diet. Some of the most promising and extensively investigated are those present in regume, cruciferous family vegetables, allium, and tea. Now, chemoprevention with edible phytochemicals should be considered as an inexpensive and readily applicable, acceptable, and accessible approach to cancer control and management for general populations. This is particularly important in considering the sluggish progress made in cancer treatment. With health-care costs being a major issue today, it would be cost-effective to promote the awareness and consumption of phytochemicals as a preventive strategy for the public. Several nutrients and nonnutritive phytochemicals are being evaluated in intervention trials to evaluate their potential as cancer chemopreventive agents for certain types of cancer. Assuming that the nature of cancer chemoprevetion requires the use of chemical substances or mixtures with little or no toxicity, it is critical to establish safety/toxicity profiles as well as efficacy for the chronic administration of any specific phytochemicals prior to use for cancer chemoprevention clinical trials
Despite significant advances in understanding the multistage carcinogenesis, little is known about the mechanism of action of the majority of chemopreventive agents at cellular and molecular levels. The chemopreventive effects most dietary phytochemicals exert are likely to be the sum of several distinct mechanisms. Given their structural diversity, it is not feasible to deduce a common mechanism that is associated with each phytochemical structure. Disruption or deregulation of intracellular signaling cascades often leads to malignant transformation of cells, and it is hence important to identify the molecules in the signaling network that can be affected by individual chemopreventive phytochemicals for better assessment of their underlying mechanisms.
Development and use of chemopreventive agents in cancer chemoprevention have a multidisciplinary scientific base. The term 'nutragenomics' has recently been coined, and much attention is being focused on this relatively new area of research. Tailored supplementation with designer foods consisting of chemopreventive phytochemicals with distinct mechanisms will be available in the future with the advances in the genetic and molecular epidemiology of carcinogenesis.
Acknowledgments
This work was supported by the National Research Laboratory (NRL) grant from KISTEP, Ministry of Science and Technology, Republic of Korea.