Tumor Microenvironment Can Predict Chemotherapy Response of Patients with Triple-Negative Breast Cancer Receiving Neoadjuvant Chemotherapy
Article information

Abstract
Purpose
Triple-negative breast cancer (TNBC) is a breast cancer subtype that has poor prognosis and exhibits a unique tumor microenvironment. Analysis of the tumor microbiome has indicated a relationship between the tumor microenvironment and treatment response. Therefore, we attempted to reveal the role of the tumor microbiome in patients with TNBC receiving neoadjuvant chemotherapy.
Materials and Methods
We collected TNBC patient RNA-sequencing samples from the Gene Expression Omnibus and extracted microbiome count data. Differential and relative abundance were estimated with linear discriminant analysis effect size. We calculated the immune cell fraction with CIBERSORTx and conducted survival analysis using the Cancer Genome Atlas patient data. Correlations between the microbiome and immune cell compositions were analyzed and a prediction model was constructed to estimate drug response.
Results
Among the pathological complete response group (pCR), the beta diversity varied considerably; consequently, 20 genera and 24 species were observed to express a significant differential and relative abundance. Pandoraea pulmonicola and Brucella melitensis were found to be important features in determining drug response. In correlation analysis, Geosporobacter ferrireducens, Streptococcus sanguinis, and resting natural killer cells were the most correlated factors in the pCR, whereas Nitrosospira briensis, Plantactinospora sp. BC1, and regulatory T cells were key features in the residual disease group.
Conclusion
Our study demonstrated that the microbiome analysis of tumor tissue can predict chemotherapy response of patients with TNBC. Further, the immunological tumor microenvironment may be impacted by the tumor microbiome, thereby affecting the corresponding survival and treatment response.
Introduction
Breast cancer is one of the most commonly diagnosed cancers and the most prevalent cause of cancer-related death in women [1,2]. Breast cancer is divided into subtypes according to expression of the estrogen, progesterone, and human epidermal growth factor 2 receptors. Triple-negative breast cancer (TNBC) accounts for roughly 15%-20% of all breast cancers and is defined as having no expression of any of the aforementioned receptors [3,4]. In particular, TNBC is prevalent in premenopausal women and those who have BRCA gene mutations [4,5]. TNBC varies from other types of breast cancer in that it has fewer treatment options, tends to have a worse prognosis, and tends to grow and spread more quickly [6]. The tumor microenvironment in TNBC exhibits a high expression of tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages (TAMs), and vascular endothelial growth factors [7,8]. CD8+ T lymphocytes, which are abundant in TNBC, possess a relationship with prognosis [9,10]. The prevalence of FOXP3+ regulatory T cells (Tregs), which can reduce anti-tumor immunity, is higher in the microenvironment than normal tissue [11,12]. Further, granulocyte colony-stimulating factor secretion, which can initiate the transformation of M1-type to M2-type macrophages, is more abundant in TNBC [13]. Overall, this unique microenvironment affects prognosis and treatment response.
In the treatment of early-stage TNBC, neoadjuvant chemotherapy (NAC) before surgery is widely used. NAC can shrink the tumor to reduce the range of surgery, improving the patients’ quality of life; additionally, this treatment exhibits similar efficacy to adjuvant chemotherapy in terms of survival and recurrence rate [14,15]. NAC can also verify if the tumor responds to the chemotherapy, and may be more beneficial than adjuvant chemotherapy because additional chemotherapy can be administered if residual tumors remain after NAC and surgery [16].
In patients receiving NAC, chemotherapy tumor response appears differently. Some patients achieve pathologic complete response (pCR) after NAC, while some others have no response or worsen. Achieving a pCR has been shown to predict clinical benefits in terms of relapse and overall survival [17]. It has been reported that 35%-45% of patients obtained pCR with the standard NAC regimen of anthracycline, cyclophosphamide, and taxane [18]. Recently, additional treatments, including immune checkpoint inhibitor or poly(ADP-ribose) polymerase inhibitors, have also been actively implemented to enhance the efficacy of NAC [19,20].
Assessment of tumor response after NAC can predict prognosis; however, if the tumor response to NAC can be anticipated before NAC treatment, it would be helpful to ascertain the most appropriate treatment options. For instance, if we can predict the response before NAC, NAC can be performed on patients with anticipated pCR, and patients who are expected to have little response may be first considered for additional treatments or surgery. However, there are currently few biomarkers or methods that can predict the response to NAC.
The microbiota, which includes bacteria, fungi, and viruses, is located in the human gut, skin, and most of the body; nonetheless, the microbiota is also found within tumors. In the past, many studies have focused on the role of the microbiome in the gut; however, recent studies on the role of the microbiome in tumor tissues have been reported. Overall, it would be important to study the correlation between the tumor microenvironment and microbiome of TNBC via microbiome analysis and determine if these correlations are related to the treatment response.
Therefore, in this study, we conducted analysis to discover potential biomarkers for predicting the response to NAC in patients with TNBC. Further, microbiome profiling conducted in the tumor tissue was performed to develop a model that could predict the corresponding tumor response to NAC.
Materials and Methods
1. Public data collection
Breast cancer RNA-sequencing data and corresponding patients’ clinical characteristics were downloaded from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA). We searched for the data from GEO using the keywords “breast cancer”, “triple-negative”, and “drug”, ultimately, obtaining the GSE163882 dataset (n=222) [21]. This data was downloaded using R GEOquery package (ver. 2.62.2 [22]). TCGA RNA-sequencing and clinical data (n=1,097) were downloaded from the Broad Institute Genome Data Analysis Center Firehose database (https://gdac.broadinstitute.org/) and Genomic Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/projects/TCGABRCA). We included TNBC and paired-end RNA-sequencing data from public databases. Processed microbial count data of TCGA breast cancer was obtained from ftp://ftp.microbio.me/pub/cancer_microbiome_analysis/ [23]. The overall process of analysis in this study is shown in Fig. 1.
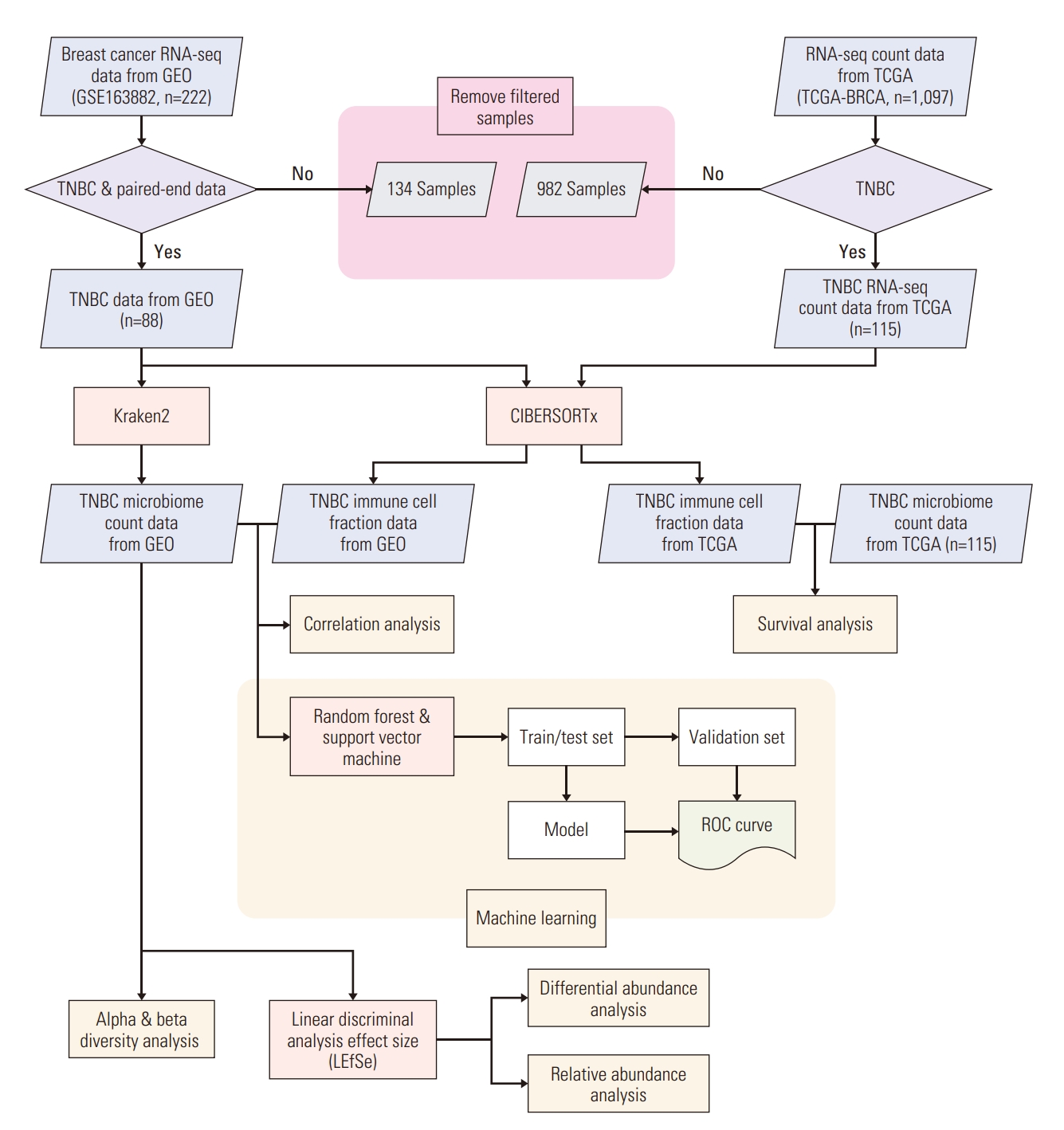
Flowchart of the analysis process in this study. GEO, Gene Expression Omnibus; RNA-seq, RNA-sequencing; ROC curve, receiver operating characteristic curve; TCGA, The Cancer Genome Atlas; TNBC, triple-negative breast cancer.
2. Analysis of the microbiome
To obtain the taxonomic classification, we used Kraken2 [24] and the Minikraken2_v1 database. Next, the Bracken [25] algorithm was used for estimating the abundance of microbiota in sample data. The microbial count data were obtained according to phylum, genus, and species level.
Processed microbial count data were divided into two subgroups: pCR and residual disease (RD). Diversity analysis was performed using a species level dataset via the phyloseq package ver. 1.38.0 [26]. Alpha diversity was calculated using the Chao1 and Shannon indices. Beta diversity was measured using Bray-Curtis dissimilarity; permutational multivariate analysis of variance (PERMANOVA) was used to examine statistical differences between the response group. Linear discriminant analysis effect size (LEfSe) was calculated using the Huttenhower Lab Galaxy Server [27] to investigate differentially abundant microbiota. LEfSe uses the Kruskal-Wallis test and linear discriminant analysis (LDA) to investigate a significant factor. A statistically notable difference was considered at a p-value < 0.05 and LDA score > 1.5.
3. Immune cell fraction analysis
Immune cell fractions of two datasets were calculated using CIBERSORTx [28], based on an autonomously provided LM22 signature matrix with 100 permutations. Quantile normalization was disabled due to the provided guideline of the tool.
4. Survival analysis
Kaplan-Meier analysis was performed by survival package (ver. 3.2-13 [29]) to analyze 5-year survival alongside TCGA’s microbiome count and immune cell fractional data. Due to the absence of drug response data in the TCGA dataset, microbial expression data and immune cell fraction values were regrouped into high and low expression groups based on the middle and mean values, respectively, of each datum’s expression.
5. Co-occurrence network
The correlation between microbiota at the species level and immune cells was analyzed by the drug response group. A Spearman’s rank correlation test, provided by the R Hmisc package ver. 4.7-0 [30], was used for analysis. Then, for those with a correlation p-value of < 0.05, a co-occurrence network was visualized using Cytoscape ver. 3.9.0 [31].
6. Machine learning
To predict the drug response using microbiota and immune cells, we built a prediction model. To build a model, we used two different types of classifiers: the support vector machine (SVM), from the e1071 package ver. 1.7-9 [32], and the random forest (RF) method, from the RF package ver. 4.7-1.1 [33] in R. We selected several features, specifically selecting taxa at the species level and immune cell subtypes and composed multiple feature sets with different up-sampling and amplification methods. Feature scaling was applied to the data when the model was trained with SVM classifiers. The train-test and validation sets were organized by classifying the data at an arbitrary 8:2 ratio. Based on the configured train-test datasets, we trained the model by selecting classifiers with a 5-fold cross-validation method; the accuracy of the trained model was measured through a separated validation set.
Although the sequencing data for microbiome assignment were not available, we obtained pertinent information regarding a gene recognized for its influence on drug response in breast cancer from the study conducted by Hatzis et al. [34]. Subsequently, we performed a correlation analysis between the microbiome and genes associated with drug response to indirectly validate our study.
Results
Detailed information on patients is provided in Table 1. No significant difference was found between the patient’s age, cancer stage, and grade (p > 0.05).
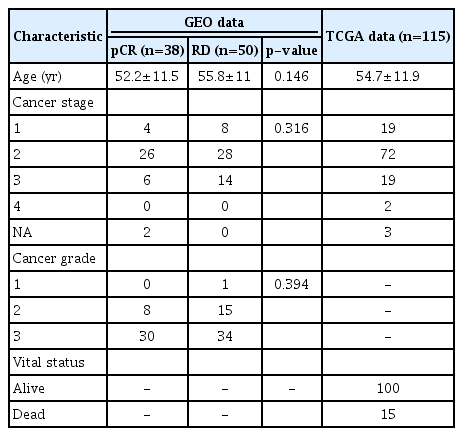
Patient characteristics in this study
1. Microbiome composition according to the drug response group
To determine overall microbial composition, we compared the relative abundance of the pCR and RD groups at the phylum and genus level (Fig. 2A). At the phylum level, we selected all the 23 phyla that showed an expression in Kraken; nonetheless, there was no big difference between the two groups. Within the genus level, we examined the top 30 taxa with high abundance, and 31 genera were used for analysis. The overall relative abundance was lower in the RD response group and the difference of several taxa, such as Pasteurella, Klebsiella, and Vibrio, was exhibited. No difference in alpha diversity was observed across both the Chao1 and Shannon indices (Fig. 2B). However, the beta diversity of the pCR and RD groups differed significantly (p < 0.05) (Fig. 2C).
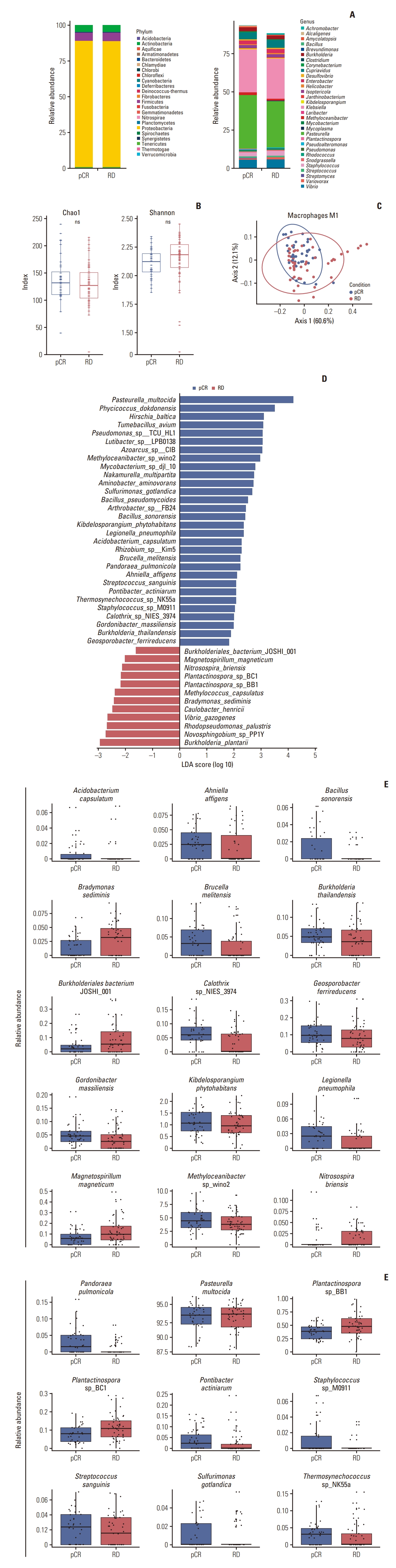
Microbiome composition of tumors from triple-negative breast cancer patients from Gene Expression Omnibus (GEO) data stratified by drug response. pCR and RD each represent pathological complete response and residual disease, respectively. (A) Stacked bar chart of the relative abundance of the microbiome at phylum and genus level; 23 phyla for the entire group and the 30 most abundant genera for each response group were evaluated. (B) Alpha diversity for all patients. Chao1 index and Shannon index were used for alpha diversity analysis. ns, not significant. (C) Beta diversity for all patients (p < 0.05). (D) Differential abundance analysis at species level using linear discriminant analysis effect size (LEfSe) stratified by drug response (linear discriminant analysis [LDA] score > 1.5, p < 0.05). (E) Relative abundance comparison for each microbiome group. The microbiome selected was not affected by outliers.
Differential abundance analysis using LEfSe was conducted to determine an enriched microbiome from each response group. At the genus level, 32 taxa were identified as significantly different (S1A Fig.). However, we assumed that some taxa may potentially appear as enriched because of outliers present; therefore, the relative abundance of each genus was additionally analyzed. As a result, 20 genera exhibited a clear difference in relative abundance (S1B Fig.). Next, we analyzed differential abundance at the species level. Consequently, 42 species exhibited a significant difference between response groups (Fig. 2D). Relative abundance analysis was also conducted at the species level; consequently, 24 species displayed a strong difference in relative abundance (Fig. 2E, S2 Fig.).
2. Predicting drug response using machine learning
A total of 18 feature sets were organized by arranging immune cell fraction and microbiome data with selected taxa from the GEO data and adjusting the application of up-sampling and amplification (S3 Table). Sets 01 and 03 were observed to express the highest accuracy in the validation process at 76.47% and 70.59% at RF and 88.24% and 82.35% at SVM, respectively. Set 01 was created with selected taxa that were dominantly expressed in the pCR group and set 03 comprised whole selected microbiome data.
We created a receiver operating characteristic (ROC) curve for each model (Fig. 3A and B). Set 01 showed a higher area under the ROC curve (AUC) in both classifiers. Additionally, the AUC for SVM was greater than RF in both datasets. When evaluating the AUC with the trained model and validation results, set 01 exhibited a higher AUC in validation at both classifiers, whereas set 03 expressed a higher AUC in the trained model.
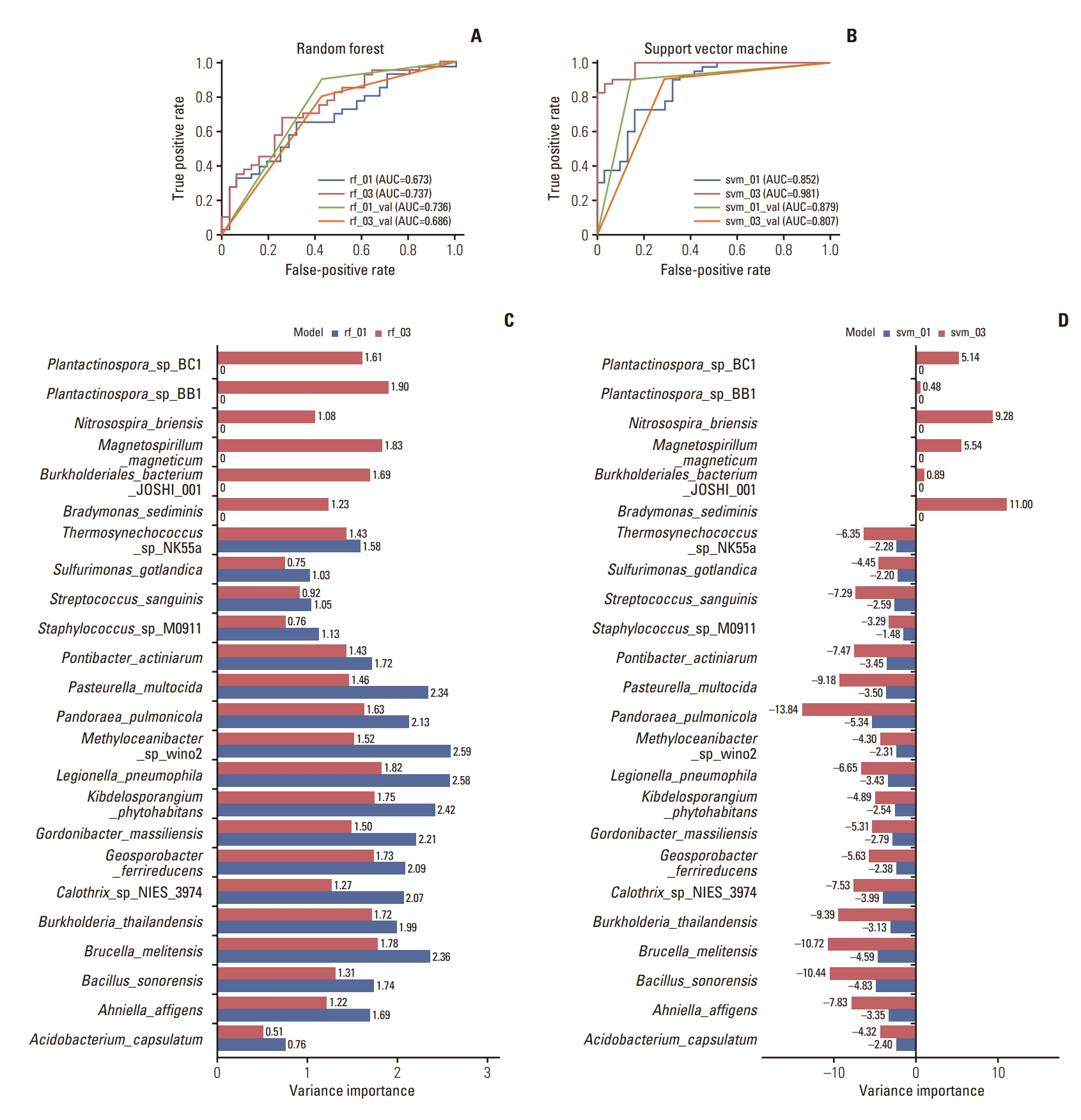
Classification model development by drug response. rf, random forest; svm, support vector machine; val, validation set. 01: dataset composed of the dominantly expressed microbiome in pathological complete response patients according to the linear discriminant analysis effect size (LEfSe) result; 03: dataset composed of the microbiome that is significantly different according to the LEfSe result. (A, B) Receiver operating characteristic (ROC) curve and corresponding area under the ROC curve (AUC) values for train-test and validation sets from the models classified with rf and svm, respectively. (C, D) Bar chart showing variance importance from the models classified with random forest (RF) and support vector machine (SVM), respectively.
To discover a feature that primarily affects drug response, we measured variable importance (Fig. 3C and D). In the SVM classifier, P. pulmonicola, Bacillus sonorensis, and Brucella melitensis were important features in both models to determine drug response. Additionally, Bradymonas sediminis was a key variable in the svm_03 model; however, RF expressed a different pattern. Legionella pneumophila was an important feature for both models; nonetheless, Methyloceanibacter sp. wino2 and Kibdelosporangium phytohabitans for rf_01, Plantactinospora sp. BB1 and Magnetospirillum magneticum for rf_03 were also important variances in determining drug response.
In order to validate our model, we performed a correlation analysis between the features of our microbiome and genes that are known to impact drug response. Our analysis revealed a positive correlation between two factors that both predict the same drug response (S4 Fig.).
3. Co-occurrence between microbiome and immune cell
To observe a co-occurrence between selected taxa and immune cell subtypes, correlation analysis was performed (Fig. 4A and B). Both response groups were analyzed individually, with immune cell fractional data of each particular patient and microbiota expressed dominantly in the group. Both groups represented a remarkable positive and negative correlation.
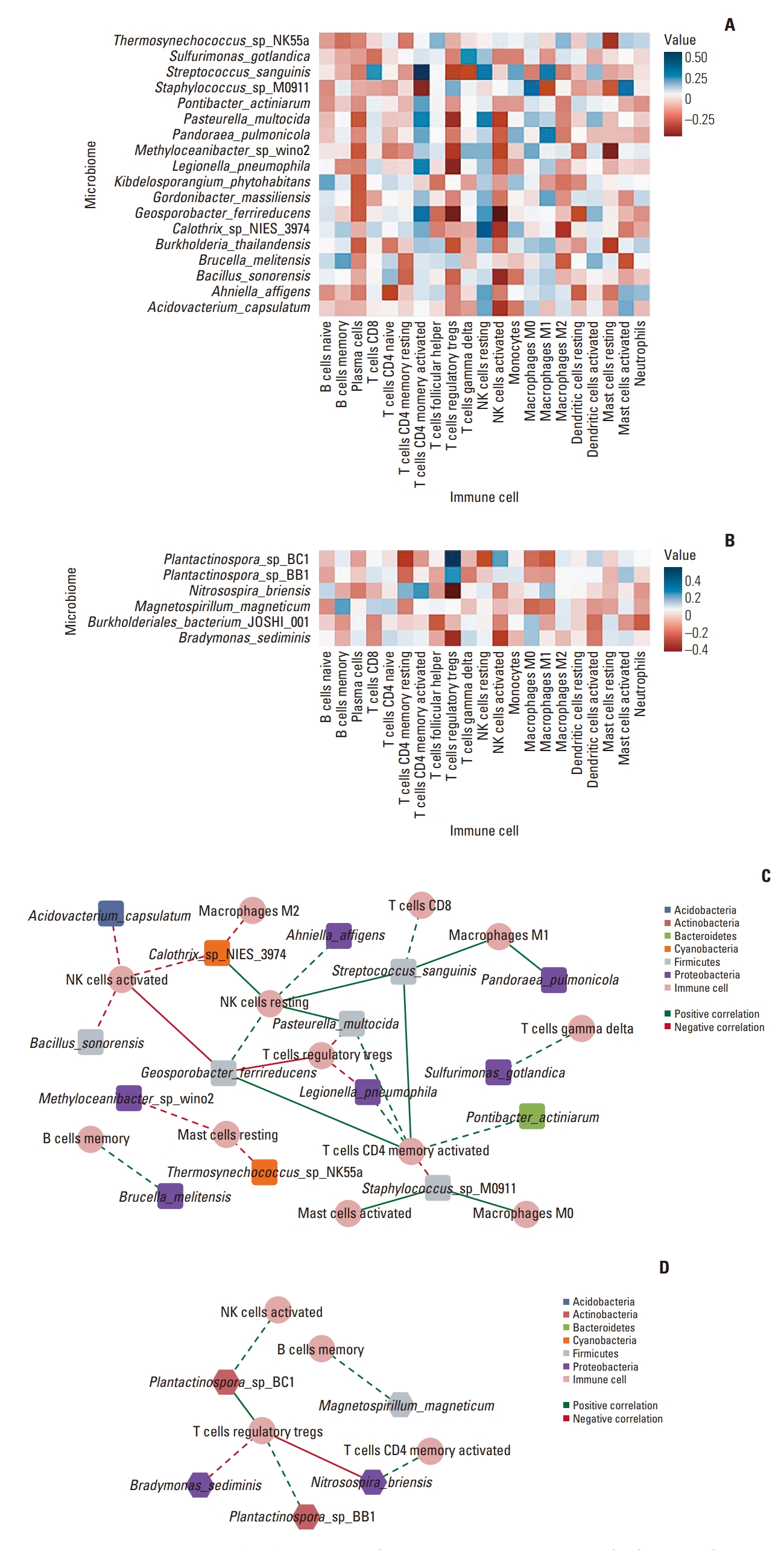
Correlations between microbiome and immune cell composition with a significant difference. (A, B) Heatmap plot indicating correlations between microbiome and immune cell composition in pathological complete response (pCR) and residual disease (RD) response patients, respectively. (C, D) network plot in the pCR and RD response, respectively. The phylum of each microbiome is marked with colors which are defined in the key. Round: immune cell, diamond: microbiome, round rectangle: microbiome dominantly expressed in the pCR patient group, hexagon: microbiome dominantly expressed in the RD patient group, dotted line: correlation coefficient (r) > 0.3, solid line: r > 0.4.
We created a network plot based on these findings, excluding edges that did not satisfy the threshold (Fig. 4C and D). This result demonstrated that the pCR group formed a more complex network than RD. In both groups, memory B cells, activated natural killer (NK) cells, activated CD4+ memory T cells, and Tregs exhibited relationships with the microbiota. Six taxa, including Geosporobacter ferrireducens, Streptococcus sanguinis, and P. pulmonicola expressed high correlation coefficients with immune cell subtypes in the pCR group. Further, resting NK cells, M1 macrophages, and activated CD4+ memory T cells exhibited high correlations with the microbiota. Alternatively, in the RD group, Plantactinospora sp. BC1 and Nitrosospira briensis possessed high correlation coefficients with Tregs.
4. Effects of immune cells and microbiome on survival
Five-year survival analysis was conducted on TCGA data with selected taxa from the GEO dataset analysis at the genus level and immune cell fraction data. M2 macrophages did not show an association with other microbiota but had a significant effect on survival, indicating an improved prognosis with lower expression (Fig. 5A). Among the immune cell subtypes that showed a correlation in both groups or high co-occurrence, M1 macrophages and resting NK cells possessed a tendency to increase the survival rate when the expression rate was high (Fig. 5B and C), whereas other immune cell subtypes did not indicate significance (Fig. 5D-I). The survival rate of the other immune cells, which did not show a correlation with microbiota, did not differ significantly (S5 Fig.). Among the selected microbiota, Brucella demonstrated a significant difference in survival rate, showing greater survival with reduced expression (Fig. 5J). However, the remaining microbiota taxa had no influence on survival (S6 Fig).
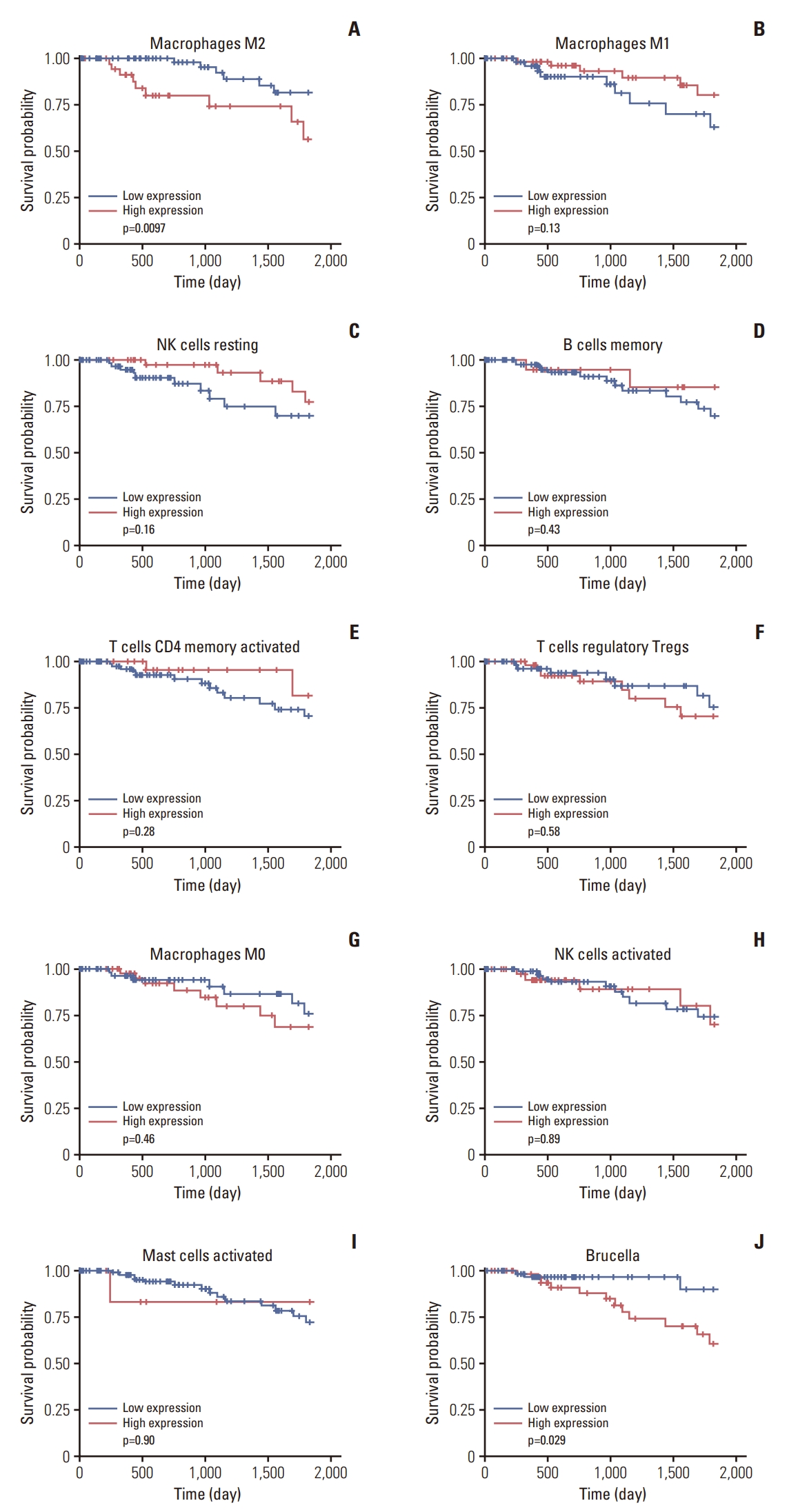
The Kaplan-Meier plot for the 5-year survival analysis using the immune cell fraction data and microbiome count data from the Cancer Genome Atlas (TCGA; n=115) triple-negative breast cancer patients: (A) macrophages M2, (B) macrophages M1, (C) NK cells resting, (D) B cells memory, (E) T cells CD4 memory activated, (F) T cells regulatory Tregs, (G) macrophages M0, (H) natural killer (NK) cells activated, (I) mast cells activated, and (J) Brucella.
Discussion
The difference in microbiome composition between tumor and normal tissues has been previously reported [35,36]. The tumor microbiome can increase genetic mutations, thereby promoting tumorigenesis, modulating the immune system, and potentially influencing cancer progression [37]. The role of the tumor microbiome on the presentation of clinical features and treatment response has been reported in several studies. Pseudomonas may reduce metastasis of prostate cancer [37], Acidovarax spp. are abundant in lung cancer patients who smoked [38], and Fusobacterium nucleatum may promote metastasis of colorectal cancer [39].
Additionally, the overall abundance of bacteria is higher in breast cancer tissues than in normal breast tissues. Further, the microbiome in breast cancer is more abundant and diverse than that of other tumors [40]. Several studies also reported the relationship between the tumor microbiome and clinical features in breast cancer. Lactobacillus was determined to be positively correlated with lymphovascular invasion and lung metastasis; similarly, F. nucleatum has been associated with tumor growth and metastatic progression [41,42].
The tumor microbiome differs among the subtypes of breast cancer, including in TNBC and non-TNBC [40]. Compared to those in non-TNBC, Alkanindiges, Caulobacter, Proteus, Brevibacillus, Kocuria, and Parasediminibacterium were determined to be enriched in TNBC [41]. These differences in the tumor microbiome may affect the tumor microenvironment. In TNBC tumor tissue, TILs and TAMs are more abundant and M2-type macrophages are up-regulated when compared to those in other breast cancer subtypes. Further, cancer-associated adipocytes in TNBC have a more pronounced effect on the migration of cancer cells and, ultimately, promote tumor progression [43]. These factors may contribute to a poorer prognosis for patients with TNBC compared to the prognosis of those with other breast cancer subtypes. Nonetheless, studies using immune checkpoint inhibitors may have only shown strong results in the TNBC breast cancer subtype because of these specific characteristics [20].
In this current study, we confirmed significant differences in the tumor microbiome according to the treatment response in patients with TNBC receiving NAC. We also found correlation of the tumor microbiome and immune cell compositions in pCR and RD groups; further, the effect of tumor microbiome and immune cells on survival was also revealed. Several microbiome including P. pulmonicola, B. sonorensis, B. melitensis, and L. pneumophila were revealed as important features in determining the response of patients to NAC. Some microbiomes showed significant correlations with specific immune cell subtypes in both groups. With low expression of M2 macrophages, a known up-regulated factor in TNBC, improved prognosis was observed; additionally, high expression of M1 macrophages and resting NK cells indicated a tendency towards improved survival chances. Overall, we found correlations between tumor microbiome composition and clinical outcome and, ultimately, determined the mechanism of the tumor microbiome in the overall tumor microenvironment. The results of this study are significant in that they can predict the treatment response of patients undergoing NAC. Therefore, this may be helpful in determining the appropriate treatment strategy for TNBC patients before surgery. By analyzing the microbiome in the biopsy tissue at the time of diagnosis, the response to chemotherapy can be predicted and it can be possible to decide what specific treatment would be most appropriate for each individual.
This study, however, has several limitations. First, we did not analyze fresh tissue. Specifically, analysis of the microbiome in fresh tissue samples may be considered to be more accurate; therefore, there are some limitations in the interpretations of these findings. Further, the results of this study were obtained via retrospective analysis of previously registered information, in place of a prospective study. Nonetheless, since the analysis in the current study exhibited significant results, it would be meaningful to verify these results through a prospective study using fresh tissue in the future.
In conclusion, the response of chemotherapy can be predicted through microbiome analysis performed on tumor tissue in patients with TNBC receiving NAC. Additionally, the tumor microbiome may affect the immune microenvironment of the tumor, which may affect treatment response and survival.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Author Contributions
Conceived and designed the analysis: Kim YH, Kim JJ.
Collected the data: Kim D, Yu Y.
Contributed data or analysis tools: Kim YH.
Performed the analysis: Kim D, Yu Y, Jung KS.
Wrote the paper: Kim D, Jung KS, Kim JJ.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgments
The result shown here are based on data from the GEO and the TCGA Research Network.
This work was supported by the Medical Research Center program [2018R1A5A2023879] from National Research Foundation of Korea, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI22C1377). This study was also supported by Research institute for Convergence of biomedical science and technology (30-2020-011), Pusan National University Yangsan Hospital.