Phenylacetate Induces Growth Inhibition and Apoptosis of Human Osteosarcoma Cells
Article information
Abstract
Purpose
Phenylacetate has potent antiproliferative effects in many malignant tumors. However, the exact mechanism as to how phenylacetate induces cell growth arrest remains unclear and very little is known about its effects on human osteosarcoma cells. In this study, we investigated whether phenylacetate is effective against two osteosarcoma cell lines (HOS and U-2 OS) in vitro.
Materials and Methods
The viability of phenylacetate-treated cell lines was assessed by trypan blue exclusion assay, and the cell cycle distribution was measured by flow cytometry. To measure cell apoptosis, poly (ADP-ribose) polymerase cleavage assay and flow cytometry were employed. The expressions of cell cycle-regulatory proteins and the apoptosis-related genes were evaluated by western blot analysis.
Results
Phenylacetate was found to inhibit the growth of osteosarcoma cells, induce cell cycle arrest in the G1 phase, and induce apoptosis. A significant decrease in Bcl-2 expression and a mild up-regulation of Bax were also observed in both phenylacetate-treated cell lines. Reduced phosphorylation of the pRb and the increased expression of p21Cip1 were observed subsequent to treatment with phenylacetate.
Conclusion
These findings support the idea that phenylacetate may be an effective chemotherapeutic agent to be employed in the future against osteosarcoma, because phenylacetate acts to inhibit the growth of osteosarcoma cells through cell cycle arrest and apoptosis.
INTRODUCTION
Osteosarcoma is a frequently seen, highly malignant tumor of the bone, and this disease exhibits a peak incidence during the second and third decades of life. Despite recent advancements in multimodality treatments, which consist of aggressive adjuvant chemotherapy and wide tumor excision, distant metastasis occurs in approximately 40~50% of patients with osteosarcoma. This distant metastasis remains a major cause of fatal outcomes (1). Effective adjuvant chemotherapy has greatly improved the long-term disease free 5-year survival rates from 10~20% in 1970s to 60~70% in recent years (2). In spite of the improved survival rate, non-responding patients still remain a major issue (3). One possible method of treatment is the use of an alternative chemotherapeutic drug as a postoperative therapeutic regimen, though this treatment requires an in vitro research validation.
Small molecules, such as aromatic fatty acids, are currently being proposed as a new class of tumor growth inhibitory compounds. Phenylacetate (PA), a physiological product of phenylalanine metabolism, is present at micromolar levels in human plasma (4), and is conjugated to glutamine in the liver by phenylacetyl coenzyme A to form phenylacetyl-glutamine.
PA has been reported to have potent antiproliferative and differentiating effects on hematologic malignancies and on many solid tumors (5~11). However, the exact mechanism as to how PA induces cell growth arrest remains unclear.
Apoptosis is one of the principal pathways by which chemotherapeutic agents inhibit the growth of cancer cells. Thus, it is necessary to investigate whether the induction of apoptosis and the alteration of apoptosis-related gene expression are important in the determination of the molecular mechanism(s) by which PA exerts its effects on osteosarcoma cells in culture. The induction of apoptosis is partially mediated by several genes such as p53, Bcl-2, and Bax (12). Among these, Bcl-2 is one of the most crucial genes involved in apoptotic processes. It functions as a suppressor of apoptotic death and is triggered by various signals. Its overexpression has been shown to inhibit apoptosis (13). It has been reported that PA induces growth inhibition, apoptosis, and Bcl-2 down-regulation in breast cancer cells in vitro, and in nude mice (14).
In addition to the apoptotic process, cell growth is also regulated through a delicate balance of positive and negative regulatory components that exert their effects during the G1 phase of the cell cycle. The most critical positive-acting components are the G1 cyclins (cyclin D and E) (15). These cyclins are assembled by cyclin-dependent kinase (CDK), and phosphorylate the key physiological substrate, retinoblastoma protein (pRb) (16). There are at least two distinct families of CDK inhibitors evident in mammalian cells: the Kip/Cip family and INK4 family. The former includes p21, p27 and p57, while the latter includes p15, p16 and p19. Members of the Kip/Cip family bind to a wide variety of cyclin/CDK complexes, preventing their activation by CDKs and their entry into the S phase. An increased expression of p21 was observed in renal cancer cell lines following treatment with PA (17). The expression of p27 had been increased by PA in human prostate cancer cells (18). It was also suggested that p27 might be a critical target in PA-mediated cell growth arrest in prostate cancer cells, and that p27 plays a key role in CDK2 inactivation, followed by hypophosphorylation of pRB, and the subsequent cell cycle arrest at the G1 phase.
PA has not yet been used on patients with osteosarcoma and very little is known about its in vitro effects on human osteosarcoma cells. The aim of this study is to test the anti-tumor effects of PA and to investigate the effects of the growth inhibition mechanism of PA on osteosarcoma cells in vitro. This will provide a basis for the possible use of PA in vivo.
MATERIALS AND METHODS
1) Materials
PA was obtained from Sigma-Aldrich, Inc. (St. Louis, MO). Antibodies against poly (ADP-ribose) polymerase (PARP), Bcl-2, Bax, pRB, and p21Cip1 were purchased from a Santa Cruz Biotechnology (Santa Cruz, CA). Propidium iodide (PI) and RNase A were purchased from Sigma Chemical Corporation (St. Louis, MO).
2) Cell culture
The human osteosarcoma cell lines, HOS and U-2 OS, were obtained from the Korean Cell Line Bank (KCLB, Korea) and cultured in RPMI 1640 (HOS) or McCoy's 5A medium (U-2 OS) containing 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml), streptomycin (100 mg/ml) in a water-jacketed incubator with a humidified atmosphere (5% CO2, 95% air) at 37℃. Experimental cultures were grown to 60~70% confluence. The cells were then treated with PA or ethanol alone by replacing the medium with fresh media containing 10% FBS and the test compound. The amount of ethanol added as the vehicle never exceeded 0.1% of the total volume.
3) Cell proliferation assay
Each cell line was seeded at 1×106 cells/ml per 100 mm on a culture plate. The culture medium was replaced with fresh medium, and PA or ethanol was added after 24 h. After 24, 48, or 72 hours, the adherent cells were detached with trypsin/EDTA, and the cell number was determined using a hemocytometer. Cell viability was assessed by trypan blue exclusion assay.
4) Colony survival assay
A colony formation assay was used to determine the long-term survival of cells following a treatment with PA (5 mM) or ethanol alone. After a short period of time, exponentially growing cells were plated at a density of 4×104 cells/ml onto 100 mm culture dishes, and the reagents were added after 24 hours. Two weeks later, the cells were fixed with methanol and stained with 3% crystal violet.
5) Cell cycle analysis
For the purposes of cell cycle analysis, the cells were plated at an initial density of 1×106 cells/ml per 100 mm onto culture plates. The culture medium was replaced with fresh medium and PA or ethanol was added after 24 hours. Forty eight hours after treatment, the cells were trypsinized, washed once with phosphate buffered saline (PBS), and fixed in 70% ethanol for at least 1 hour at 4℃. After washing with PBS, they were incubated at room temperature with PI staining solution (0.005 mg/ml propidium iodide, 20 µg/ml RNase A) for 1 hour in the dark. Stained cells were analyzed by FACScan (Becton Dickinson, Mansfield, MA). The percentages of the cells in the G0/G1, S and G2/M phases were determined by cell Quest software (Becton Dickinson, Mansfield, MA).
6) Western blot analysis
The cell extracts were prepared as follows. Cells were collected by centrifugation and dissolved in a cold lysis buffer [1×PBS, 1% Triton X-100, 0.05% Sodium deoxycholate, 0.0l% SDS, 0.5 µg/ml leupeptin, l mM EDTA, 1 µg/ml pepstin and 0.2 mM PMSF] for 30 minutes on ice. The concentrations of total proteins in the extracts were determined using the Bradford protein assay. Bovine serum albumin (BSA) was used as a protein standard. Cell extracts (30 µg of protein/lane) were separated on 6~12% SDS-polyacrylamide gels with the Tri glycine buffer system, and the proteins were transferred to polyvinyliden difluoride (PVDF) membranes (Boehringer Mannheim, Germany) using a semi-dry transfer system as recommended by the manufacturer (Bio-Rad, Hercules, CA). After blocking with PBST (phosphate buffered saline pH 7.4, 0.1% Tween 20) containing 5% low fat milk, the membranes were incubated with the primary antibody in PBST for 1 hour. The membranes were then washed twice in PBST for 10 minutes. Next, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase (HRP) for 1 hour and developed with the enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech, UK). Equal protein loading was controlled by a reprobing process with anti-actin antibody (1:1,000, Sigma-Aldrich, Inc. St. Louis, MO)
RESULTS
1) Effect of PA on cell growth
Growth inhibition studies were performed using osteosarcoma cell lines HOS and U-2 OS. These cells were tested with increasing nontoxic concentrations of PA at 2, 5, 7, 10 and 15 mM. As shown in Fig. 1, composite inhibitory effects exist at different concentrations of PA after 24, 48, and 72 hours of exposure. The antiproliferative effect of PA was time- and dose-dependent in both cell lines. The inhibitory effects of PA were not due to nonspecific cytotoxic action because cell viability, as assessed by trypan blue exclusion, was greater than 95%. This did not differ between control and treated cells. Both the HOS and U-2 OS cells, each treated with 5 mM of PA, failed to form colonies after 2 weeks on the colony survival assay (Fig. 2). The inhibition of cell proliferation could have been due to the induction of apoptosis elicited by PA. Therefore, further studies were performed to investigate whether PA could induce apoptosis in osteosarcoma cells.
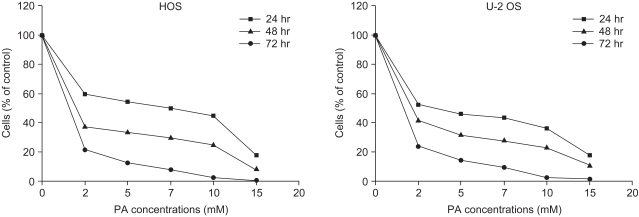
In the study of the effect of PA on growth of osteosarcoma cells, PA had a time- and dose-dependent antiproliferative effect in both osteosarcoma cell lines (HOS and U-2 OS).
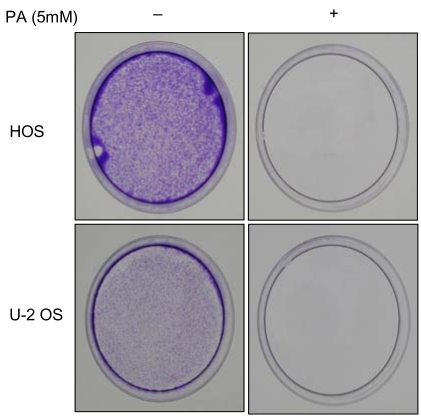
In the study of the effect of PA on the long-term survival of osteosarcoma cells, HOS and U-2 OS cells treated with 5 mM of PA failed to form colonies, as measured at 14 days after treatment. Both lines of osteosarcoma cells were treated with ethanol as a control.
2) Induction of cell cycle arrest and apoptosis
Flow cytometric analysis revealed that PA treatment resulted in a higher proportion of cells in the G0/G1 phase relative to the control cells (HOS: 55% in control and 62.3% in treated cells, exclusive of apoptotic cells; U-2 OS: 49% in control and 63.3% in treated cells, exclusive of apoptotic cells). In contrast, PA treatment reduced the proportion of cells in the S and G2/M phase for both HOS and U-2 OS cells (Fig. 3). These results indicate that growth inhibition of osteosarcoma cells subsequent to PA treatment can be attributed to cell arrest in the G1 phase.
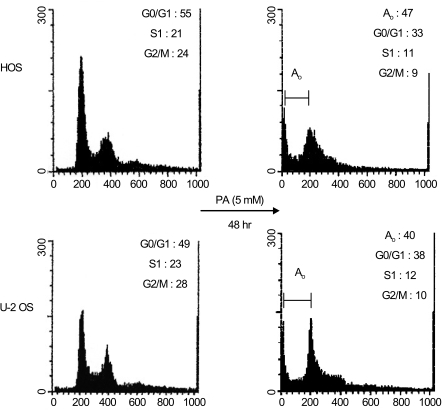
On the analysis of DNA content by flow cytometry, HOS and U-2 OS cells displayed apoptosis when treated with 5 mM of PA for 48 hours.
Using flow cytometric analysis, apoptosis was demonstrated in both the HOS and U-2 OS cells treated with 5 mM of PA. As shown in Fig. 3, 47% of HOS cells and 40% of U-2 OS cells are present in the sub-G0/G1 apoptotic phase after 48 hours of exposure to PA. The induction of apoptosis was first detected after 24 hours of treatment. This is directly correlated with the inhibition of cell proliferation. Western blot analysis revealed that after treatment with PA, the PARP protein (115 kDa) was degraded to an accumulation of the cleaved PARP (85 kDa) (Fig. 4). These two independent methods of measuring apoptosis have provided strong evidence that apoptosis was induced in both kinds of osteosarcoma cells treated with PA. In order to explore the mechanisms by which PA induces apoptosis, the alterations in the expression of Bcl-2 and Bax genes, known to be involved in the apoptotic processes, were investigated.
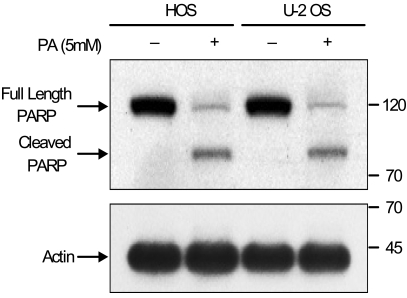
HOS and U-2 OS cells undergoing apoptosis after treatment with PA showed the cleavage of PARP by the use of Western blot analysis. This change of PARP processing is demonstrated by the progressive disappearance of the full length PARP (115 kDa) and by the progressive appearance of the cleaved PARP (85 KDa). The blots were probed with β-actin to normalize each lane for protein content.
3) Effects of PA on Bcl-2 and Bax
The effects of PA on the expression of Bcl-2 and Bax in osteosarcoma cells were studied by employing Western blot analysis. A significant decrease in Bcl-2 expression was observed in both HOS and U-2 OS cells after treatment with PA for 48 hours. However, the level of Bax expression in the osteosarcoma cells was mildly up-regulated (Fig. 5).
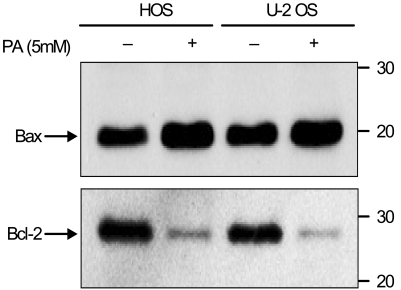
HOS and U-2 OS cells treated with 5 mM of PA for 48 hours showed the down-regulation of Bcl-2 and the up-regulation of Bax expression by using Western blot analysis. Both lines of osteosarcoma cells were treated with ethanol as a vehicle control.
4) Effects of PA on pRB phosphorylation and p21Cip1
The phosphorylation pattern of pRb is an established indicator of the proliferative status of the cell. Hypophosphorylated pRb is found in G1 cells, whereas hyperphosphorylated pRb is characteristic of proliferating cells in the S-, G2- and M phases. As shown in Fig. 6, exposure of the cells to PA resulted in a marked decrease in the expression of phosphorylated pRb.
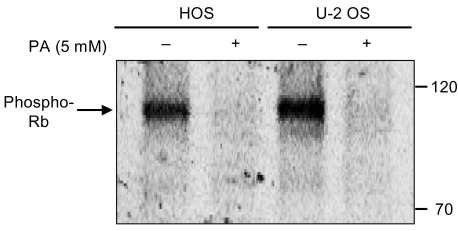
In the study of the effect of PA on pRb in osteosarcoma cells, the expression of phosphorylated pRb decreased markedly in the HOS and U-2 OS cells treated with 5 mM of PA for 48 hours. Both osteosarcoma cell lines were treated with ethanol as a control.
For negative cell cycle regulators, the expression of p21Cip1 was investigated by employing Western blot analysis. Both of the osteosarcoma cell lines showed a significant increase in p21Cip1 levels after 48 hours of exposure to 5 mM of PA (Fig. 7). The expression of p27 was not investigated.
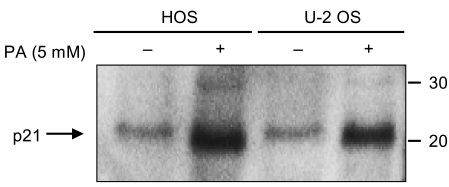
In the study of the effect of PA on p21Cip1 in osteosarcoma cells, the expression of p21Cip1 increased significantly in HOS and U-2 OS cells treated with 5 mM of PA for 48 hours. Both osteosarcoma cells were treated with ethanol as control.
DISCUSSION
The purpose of this study was to investigate the mechanism of PA-induced cell growth arrest and to evaluate the possibility of the therapeutic use of PA for osteosarcoma. It has been established that PA has an anti-proliferative effect for prostate carcinoma (6), breast cancer (7), pancreatic adenocarcinoma (8), chronic lymphocytic leukemia (9), ovarian carcinoma (10), and in the medulloblastoma and astrocytoma cell lines (11). Consistent with other studies, our results have also shown that cell growth inhibition occurred at clinically achievable concentrations of PA (2~15 mmol/L). A number of mechanisms have been proposed to explain the anti-tumor actions of PA: a) depletion of circulating glutamine, upon which cancer cells are highly dependent for nucleic acid and protein synthesis (19); b) inhibition of the protein prenylation and glycosylation, and most notably, that of Ras and lamins (20); c) hypomethylation of DNA (21); and d) activation of the peroxisomal proliferator-activated nuclear receptor (PPAR), a transcription factor that has been shown to regulate the expression of genes which control lipid metabolism (22). However, the ways in which these proposed mechanisms interact in regards to cell growth arrest and differentiation by PA and its potential molecular mediators are not well understood.
In this study, the results of flow cytometric analysis indicated that growth inhibition of osteosarcoma cells after PA treatment was due to apoptosis and cell cycle arrest. Through flow cytometric analysis, we found that 47% of HOS cells and 40% of U-2 OS cells underwent apoptotic cell death under the influence of PA treatment. Another method we used to detect apoptosis was the cleavage of PARP, which was used as an early marker of apoptosis. PARP cleavage was observed in the osteosarcoma cells treated with PA for 48 hours. This clearly suggests that these results in regards to PA-induced apoptosis are consistent with another report showing that apoptosis was induced in PA-treated breast carcinoma cells, in this case done using a TUNEL reaction-based in situ cell death detection kit (14).
Bcl-2 has been demonstrated to play a major role in the determination of whether cells will undergo apoptosis under experimental conditions which ordinarily promote cell death. Bcl-2 has been shown to protect cells from apoptosis (13). Because Bcl-2 is a major gene related to apoptosis, it is important to measure Bcl-2 expression in cancer cells treated with therapeutic agents to determine the status of cell apoptosis. In our study, a remarkable decrease in Bcl-2 expression was observed in osteosarcoma cells after treatment with 5 mM of PA for 48 hours. Another gene linked to apoptosis, Bax, promotes apoptosis. The expression of Bax in osteosarcoma cells increased slightly when treated with PA. These results were found to be consistent with the cleavage of PARP and induction of apoptosis.
Flow cytometric analysis revealed that PA treatment resulted in a higher proportion of cells in the G0/G1 phase relative to the control cells. This signifies that the growth inhibitory effects of PA are achieved when PA blocks the cell transition from the G0/G1 to the S phase of the cell cycle with a consequent recruitment of the cells in the quiescent phases of the cell cycle. These results are also consistent with other reports that have shown that an increased proportion of cells in the G0/G1 phase were induced in PA-treated renal carcinoma cells (17) and prostate carcinoma cells (18).
pRb was found to be a key regulator of the cell cycle (16). The loss of pRb's function may be responsible for uncontrolled cell cycle progression. This ultimately leads to carcinogenesis or tumor progression. According to a well-established model, active (hypophosphorylated) pRb sequesters transcription factor members of the E2F family, preventing the transcription of key genes that are critical for proliferation (23). In proliferating cells, the phosphorylation of the pRb by turns increases and decreases in every cycle: it rises late in G1 and remains high in S and G2. pRb then falls back to a dephosphorylated state when the cell goes through mitosis. As expected, the expression of phosphorylated pRb decreased markedly in both of the cell lines which had been treated with 5 mM of PA. Other studies regarding the expression of pRb in PA-treated renal cancer cells (17) and prostate cancer cells (18) have been found. These studies have reported a decrease of phosphorylated pRb compared to the control cells. Their results are similar to ours.
In most cases, the physiological role of p21Cip1 has been linked to inhibition of cyclin E/CDK2 (24). Thus, p21Cip1 is, at least in part, an essential component of the pathway for blocking the cell cycle in the G1 phase. A small number of reports in the literature explore the expression of p21Cip1 in PA-treated cancer cells. In renal carcinoma cells (17) and breast carcinoma cells (25) treated with PA, the up-regulation of p21Cip1 was observed. However, in another study with human prostatic carcinoma cells following a treatment with PA, the expression of p21Cip1 was not affected (18). In our study, the expression of p21Cip1 was increased by a PA treatment in both HOS and U-2 OS cells. This suggests that p21Cip1 is an important cyclin-dependent kinase inhibitor, possibly explaining the anti-proliferative effect of PA. Here, we can say that PA treatment to osteosarcoma cells led to substantial loss of phosphorylated pRb due, at least in part, to the inhibition of the pRb kinases (CDKs) through the increased expression of p21Cip1.
CONCLUSIONS
Our results have demonstrated that PA treatment inhibited the growth of osteosarcoma cells. This process was regulated by the expression of apoptosis-related Bcl-2 and Bax genes, and the treatment induced cell apoptosis. In addition, PA treatment decreased the expression of phosphorylated pRb, increased the level of p21Cip1, and ultimately, led to cell cycle arrest. In the near future, PA may be of great value as an effective chemotherapeutic agent against osteosarcoma. However, much remains to be studied regarding the molecular mechanisms of PA as an anti-cancer agent for osteosarcoma.
Notes
This paper was supported the Fund of Chonbuk National University Hospital Research Institute of Clinical Medicine.