HIF-1α: a Valid Therapeutic Target for Tumor Therapy
Article information
Abstract
Hypoxia plays a major role in the induction of angiogenesis during tumor development. One mechanism by which tumor cells respond to a reduced oxygen level is via the activation of hypoxia-inducible factor-1 (HIF-1). HIF-1 is an oxygen-dependent transcriptional activator that plays crucial roles in the angiogenesis of tumors and mammalian development. HIF-1 consists of a constitutively expressed HIF-1β subunit and the highly regulated HIF-1α subunits. The stability and activity of HIF-1α are regulated by various post-translational modifications, hydroxylation, acetylation, phosphorylation and sumoyaltion. Therefore, HIF-1α interacts with several protein factors including PHD, pVHL, ARD-1, SUMO and p300/CBP. Under normoxia, the HIF-1α subunit is rapidly degraded via the von Hippel-Lindau tumor suppressor gene product (pVHL)-mediated ubiquitin/proteasome pathway. The association of pVHL and HIF-1α under normoxic conditions is triggered by the hydroxylation of prolines and the acetylation of lysine within a polypeptide segment known as the oxygen-dependent degradation (ODD) domain. On the contrary, under the hypoxia condition, the HIF-1α subunit becomes stable and interacts with coactivators such as p300/CBP to modulate its transcriptional activity. Under hypoxic conditions, HIF-1 eventually acts as a master regulator of numerous hypoxia-inducible genes. The target genes of HIF-1 are especially related to angiogenesis, cell proliferation and survival, and to glucose and iron metabolism. Moreover, it was reported that the activation of HIF-1α is closely associated with a variety of tumors and oncogenic pathways. Hence, the blocking of HIF-1α itself or the blocking of HIF-1α interacting proteins inhibits tumor growth. Based on these findings, HIF-1 can be a prime target for anticancer therapies. Therefore, this review summarizes the molecular mechanism of HIF-1α stability, the biological functions of HIF-1 and its potential applications for cancer therapies.
INTRODUCTION
Hypoxia is a reduction in the normal level of tissue oxygen tension, and it occurs during several pathophysiological processes including tumorigenesis. Hypoxia may occur during the initial avascular phase or it may develop in established tumors as a result of new blood vessels formation that is ineffective and provides only poor blood flow. Hence, the growth of tumors is restricted by limited oxygen and nutrients when they are too distant from any nearby vessels. In order to overcome these restrictions, the tumors acquire the ability to recruit their own blood supply system under hypoxic stress (1). Although hypoxia generates an unfavorable situation for cell growth, cancer cell undergo a series of genetic and metabolic changes that allow them to survive and even proliferate.
The hypoxia-inducible factor-1 (HIF-1) is the most important factor involved in the cellular response to hypoxia, and it has been extensively studied during this last decade (2,3). In this review, we will summarize the oxygen dependent regulation of HIF-1 stability and function, and also its chemical implication in the field of anticancer therapies.
Hypoxia-inducible Factor (HIF); Ringmaster of the Hypoxic World
1) Structure of HIF-1
HIF-1 has been shown to be a heterodimeric consisting of an α subunit and a β subunit, and a β subunit is also known as the aryl hydrocarbon receptor nuclear translocator (ARNT). HIF-1α is the subunit that is highly regulated by the oxygen level, but HIF-1β is the constitutively expressed subunit (4). Each subunit has the basic helix-loop-helix (bHLH) and the PER-ARNT-SIM (PAS) domain. The N-terminal half of HIF-1 α contains the bHLH and PAS domains that are required for dimerization and DNA binding (5). The C-terminal half contains the domains that are required for degradation and transactivation: the oxygen-dependent degradation (ODD) domain, which confers oxygen dependent instability, two independent transactivation domains (N-TAD and C-TAD), and an inhibitory domain (ID) that negatively regulates the transactivation domains (6). Whereas the N-terminal TAD (N-TAD) also constitutes a degradation box, the C-terminal TAD (C-TAD) functions in a strictly hypoxia-inducible fashion. The C-TAD interacts with a coactivator such as p300/CBP, it is independent of protein stability and is required for full HIF activity (7) (Fig. 1).
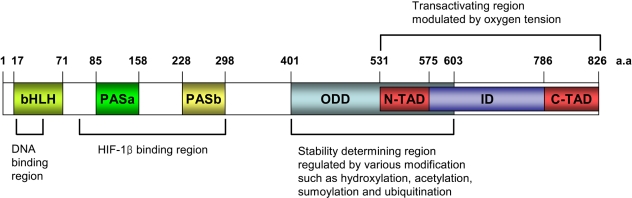
Domain structures of HIF-1α and their potential function. HIF-1α possesses the basic helix-loop-helix (bHLH) and PER-ARNT-SIM (PAS) domains that are involved in dimerization with HIF-1β and DNA binding. Its C-terminal part contains two transacting domains (TAD) and an inhibitory domain (ID). The N-TAD lies within its ODD domain. The ODD domain regulates the stability of HIF-1α via recognition by the von Hippel-Lindau E3 ubiquitin ligase (pVHL).
2) HIF-α isoforms
Homology searches in the gene bank and cloning experiments have found other members of bHLH-PAS superfamily: HIF-2α, which is referred to as endothelial PAS domain protein 1 (EPAS1), and also HIF-3α (8). These isoforms have structural similarity and are classified as members of the bHLH-PAS family. The structure and functions of HIF-1α have been more extensively studied than those of HIF-2α or HIF-3α. Six isoforms of HIF-1α have been reported. HIF-1αFL is similar to the wild type HIF-1α except for an additional three base pairs, TAG, that are between exon 1 and exon 2. Next, HIF-1α736 loses exon 14, and so it lacks the C-TAD. HIF-1αFL and HIF-1α736 activate the VEGF promoter upon hypoxia (9). In contrast to HIF-1αFL and HIF-1α736, HIF-1α557 and HIF-1α516 function as the dominant negative isoforms of HIF-1α. HIF-1α557 loses exon 12, which is induced by the zinc ion, and HIF-1α516 lacks exon 11 and 12 (10,11). Recent studies have reported that the isoform HIF-1α785 contains all the functional domains; therefore, HIF-1α785 acts as a transcriptional activator, but it lacks exon11 in the ODD domain (12). Another HIF-1α variant, HIF-1α417, conserves only the bHLH and PAS domains, which are essential for the binding between DNA and ARNT. However it lacks all the other important domains of HIF-1α, such as the ODD domain and TAD (13). Not much detail is known about the roles of the 2α and 3α class subunits. Like HIF-1α, HIF-2α is also regulated by enzymatic hydroxylation of the conserved proline residue that causes its degradation under normoxia conditions via the ubiquitin E3 ligase complex (14~17). It has recently been reported that HIF-3α also undergoes degradation through the polyubiquitination-proteasome pathway (18). Human HIF-3α has multiple spliced variants: HIF-3α1~6 (Fig. 2). HIF-3α1, 2 and 3 share a common ODD that includes the consensus motif of proline hydroxylase and it binds the pVHL E3 ligase complex under normoxic conditions. Dominant negative regulator of HIF-α function, IPAS, is generated by alternative splicing of the HIF-3α locus (19). IPAS prevents the interaction of HIF-1α with HIF-1β because IPAS dimerizes HIF-1α, and the IPAS/HIF-1α complex does not bind to the hypoxia-response element. So it inhibits transcriptional activation of HIF-1α. In contrast to HIF-3α, HIF-2α activates transcription and induces the hypoxia-mediated gene expression of such factors as VEGF. It was reported that hypoxia did not affect the mRNA levels of HIF-1α, HIF-1β and HIF-2α, but HIF-3α mRNA was increased after 2 hr of hypoxia (20). However, how the hypoxia increases the mRNA level of HIF-3α is not known. Therefore, further study concerning the expression and stability of HIF-3α is required.
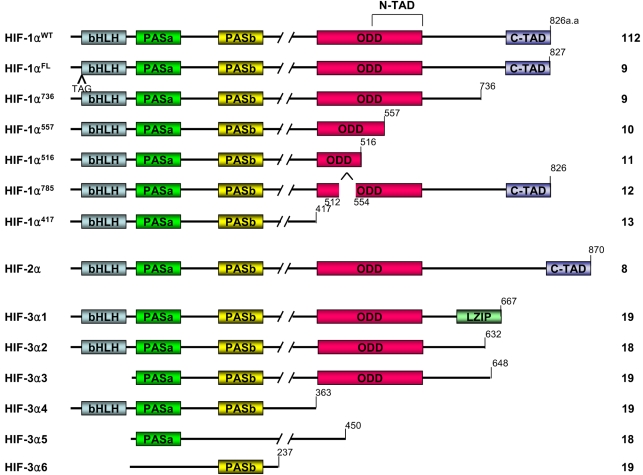
Splice variants of the HIF α subunit. bHLH: basic helix-loop-helix, PAS: Per/Arnt/Slim domain, ODD: oxygen dependent degradation domain, N-TAD: N-terminal transactivation domain, C-TAD: C-terminal transactivation domain, LZIP: leucine zipper.
The effect of the HIF family on gene expression may be different according to the cell types. For example, HIF-1α and -2α are abundantly expressed in the kidney. Nevertheless, the overexpression of HIF-2α, but not HIF-1α, promotes the growth of renal carcinoma cells (21~23). However, in the breast cancer cell line, HIF-1α is the major isoform required for the induction of hypoxic genes (24).
3) HIF-1 target genes
HIF-1 activity leads to the upregulation of genes that are involved in many aspects of cancer progression, angiogenesis, cell survival, glucose metabolism and invasion. More than 60 putative HIF-1 target genes have currently been identified (Fig. 3). HIF-1 targets the genes that are particularly relevant to cancer, and these genes encode angiogenic factors, glucose transporters and glycolytic enzymes, survival factor and invasion factor (25).
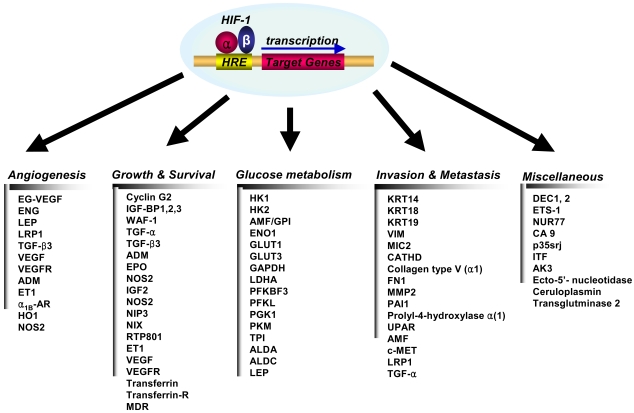
Target genes that are transcriptionally activated by HIF-1α1B-AR: α1B-adrenergic receptor, ADM: adrenomedullin, AK3: adenylate kinase 3, ALDA: aldolase A, ALDC: aldolase C, AMF: autocrine motility factor, CA9: Carbonic anhydrase 9, CATHD: cathepsin D, EG-VEGF: endocrine-gland-derived VEGF, ENG: endoglin, ET1: endothelin-1, ENO1: enolase 1, EPO: erythropoietin, FN1: fibronectin 1, GLUT1: glucose transporter 1, GLUT3: glucose transporter 3, GAPDH: glyceraldehyde-3-P-dehydrogenase, HK1: hexokinase 1, HK2: hexokinase 2, IGF2: insulin-like growth-factor 2, IGF-BP1: IGF-factor-binding-protein 1, IGF-BP2: IGF-factor-binding-protein 2, IGF-BP3: IGF-factor-binding-protein 3, ITF: intestinal trefoil factor, KRT14: keratin 14, KRT18: keratin 18, KRT19: keratin 19, LDHA: lactate dehydrogenase A, LEP: leptin, LRP1: LDL-receptor-related protein 1, MDR1: multidrug resistance 1, MMP2: matrix metalloproteinase 2, NOS2: nitric oxide synthase 2, PFKBF3: 6-phosphofructo-2-kinase/fructose-2:6-biphosphatase-3, PFKL: phosphor-fructo kinase L, PGK 1: phosphoglycerate kinase 1, PAI1: plasminogen-activator inhibitor 1, PKM: pyruvate kinase M, TGF-α: transforming growth factor-α, TGF-β3: transforming growth factor-β3, TPI: triosephosphate isomerase, VEGF: vascular endothelial growth factor, UPAR: urokinase plasminogen activator receptor, VEGFR2: VEGF receptor-2, VIM: vimentin.
(1) Angiogenesis
Vascular endothelial cell growth factor, (VEGF), is the most potent endothelial-specific mitogen to directly participate in angiogenesis (26~29), and it is one of the major target genes for HIF-1. This growth factor interacts with its receptor, VEGFR, which is specifically expressed in endothelial cells, and this stimulates endothelial cell proliferation (27,28,30,31). It has also been shown that hypoxia induces the expression of VEGF mRNA and protein, suggesting that hypoxia is a stimulus for angiogenesis through the up-regulation of VEGF expression (27,28,30,31). The induction of angiogenesis leads to an increase in the vascular density and hence, a decrease in the oxygen diffusion distance. However, local blood flow under pathophysiological conditions is controlled by modulation of the vascular tone through the production of NO (inducible nitric oxide synthase), CO (heme oxygenease 1), endothelin 1, adrenomedulin or the activation of the α1B-adrenergic receptor, and all of theseinvolve the HIF-1 target genes (32~37). Therefore, HIF-1 contributes to angiogenesis by far more complex mechanisms than simple VEGF induction, and HIF-1 probably works by recruiting additional target genes that are involved in vessel maturation (38).
(2) Growth/survival
Hypoxia-induced growth factors are known to promote cell proliferation and survival. Several growth factors, most notably insulin-like growth factor-2 (IGF2) and transforming growth factor (TGF)-α, are also HIF-1 target genes (39,40). Binding of these factors to their cognate receptors-the insulin-like growth factor 1 receptor (IGFIR) and epidermal growth factor receptor (EGFR), respectively-activates signal transduction pathways that lead both to HIF-1α expression and to cell proliferation and survival (3). The p42/p44 mitogen-activated protein kinases, which regulate cell proliferation in response to extra-cellular growth factors, have been shown to phosphorylate HIF-1α and activate transcription of HIF-1 target genes (27). Phosphatidylinositol 3-OH kinase (PI3K) activity is also increased in some cell types under hypoxic conditions (41). PI3K is one of the key downstream mediators of many tyrosine kinase signaling pathways, and it is involved in regulating cell proliferation and suppression of apoptosis. The PI3K pathway is inhibited by the phosphoinositide phosphatase PTEN, and mutations in PTEN enhance HIF-1 activated responses (42). PTEN regulates cell growth and proliferation, and it is deleted or mutated in several human cancers, including glioblastoma, endometrial tumors and prostate cancer (43). Therefore, HIF-1 contributes to the autocrine-signaling pathways that are crucial for cancer progression.
(3) Glucose metabolism
Depletion of oxygen changes the energy metabolism of cells: the cells don't use the oxygen-dependent metabolic pathway such as the TCA cycle. Instead of the TCA cycle, the cells switch to the oxygen-independent metabolic pathway, and they start using glycolysis as the primary mechanism of ATP production (44,45). The TCA cycle provides 38 ATPs from glucose, but glycolysis provides only two. Therefore, the hypoxic condition requires more glycolysis than normoxic condition. Many genes involved in glucose uptake and glycolysis have been identified as HIF-1 target genes (46). HIF-1 regulates the expression of all enzymes in the glycolytic pathway, as well as expression of the glucose transporters GLUT1 and GLUT3 that mediate cellular glucose uptake (47). Enhanced lactate production and hence, a decrease in pH, results from the increase in anaerobic glycolysis, and this potentially limits this source of ATP despite a sufficient glucose supply. Transmembrane carbonic anhydrases were reported to be HIF-1 target genes (48), and in fact, increased glycolysis is a normal response to proliferation: moreover, migrating cells also use this pathway as an energy source (49). The intermediary metabolites of the glycolytic pathway provide the precursors for synthesis of glycine, serine, purine, pyrimidine and phospholipids, all of which are essential for cell growth and the maintenance of cells that are under stressful conditions (43).
(4) Invasion and metastasis
Hypoxia unleashes the invasive and metastatic potential of tumor cells. HIF-1 regulates the expression of genes encoding cathepsin D, matrix metalloproteinase 2, urokinase plasminogen activator receptor (uPAR), fibronectin 1, keratins 14, 18 and 19, vimentin, transforming growth factor alpha, and autocrine motility factor, and these are all proteins that play established roles in the pathophysiology of invasion (3,39).
Molecular mechanism of HIF-1α stability
The first regulator of HIF-1 is oxygen, and HIF-1α appears to be the specific HIF-1 subunit regulated by hypoxia. HIF-1α protein is subject to rapid degradation at normoxia by the process of the pVHL-mediated ubiquitin-proteasome pathway, whereas hypoxia blocks this degradation and leading to accumulation of HIF-1α protein (50,51). The association of HIF-1α with pVHL is triggered by the post-translational hydroxylation of a proline residue that is mediated by prolyl hydroxylase (PHD) or HIF prolyl hydroxylase (HPH). The HIF-1α contains two sites for hydroxylation, Pro 402 and Pro 564, within its ODD domain and each site contains a conserved LXXLAP motif (15). The hydroxyproline residue becomes buried within the hydrophobic core of pVHL, the von Hippel-Lindau tumor suppressor protein, which is a part of the ubiquitin ligase protein complex (52,53).
All three paralogs of the HPH/PHD family have been discovered and named as follows: HPH-1/PHD-3, HPH-2/PHD-2 and HPH-3/PHD-1. When overexpressed as a tagged protein, HPH-2/PHD-2 resides in the cytoplasm, whereas HPH-3/PHD-1 is predominantly observed in the nucleus (54,55). Berra et al. (2003) proposed that HPH-2/PHD-2 is primarily responsible for HIF hydroxylation in the normoxic condition.
The prolyl hydroxylase is dioxygenase requiring oxygen and 2-oxoglutarate as a substrate. This enzyme transfers one oxygen atom to the proline residue, and the second oxygen atom reacts with 2-oxoglutarate to generate succinate. The activity of PHD to HIF-1α is known to depend on the O2 concentration. Therefore, PHD has been suggested to be an oxygen sensor (56,57).
Another protein interacting with the ODD domain of HIF-1α is ARD1 acetyltransferase (58). The yeast homolog of ARD-1 is required for the expression of protein N-acetyltransferase (NAT) in lower eukaryotes and bacteria, but the function of NAT has not been defined in mammalian cells (59,60). Jeong et al. (2002) reported that ARD1 acetylates the Lys-532 residue in the ODD domain of HIF-1α by transferring an acetyl group from Ac-CoA. The acetylation level of HIF-1α gradually decreases as the length of hypoxic exposure time increases, which is due to the reduced expression of ARD1. The acetylation of Lys532 by ARD1 is critical to the proteasomal degradation of HIF-1α. A K532R mutant that was not acetylated by ARD1 was stabilized and it showed a decreased interaction with pVHL (58). It is interesting that Lys532 was previously reported as being critical for the degradation of HIF-1α under the normoxic condition (12,61). Although it is unclear how the acetylation of HIF-1α leads to its decreased stability, a conformational change of HIF-1α may effectively increase its interaction with pVHL and enhance the subsequent proteasomal degradation.
HIF-1α is also sumoylated by SUMO-1, which is the small ubiquitin-like protein family. This modification leads to an increase in HIF-1α stability and an increase in its transcriptional activity, implying that SUMO-1 may counteract ubiquitin. We recently reported that sumoylation occurs at Lys391/Lys477 residues, but not at the Lys532 residue (Fig. 4)(62)
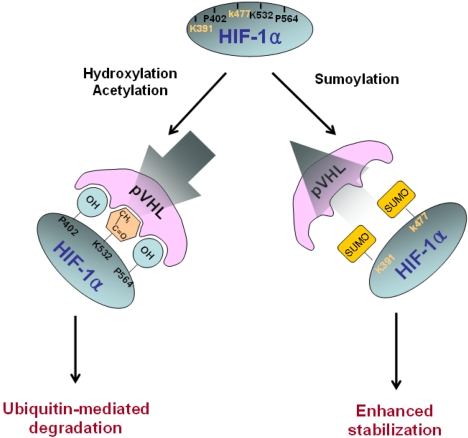
Molecular mechanism of HIF-1α stability. HIF-1α is subject to rapid degradation at normoxia by the pVHL-meditated ubiquitin-proteasome pathway, whereas hypoxia blocks degradation of HIF-1α leads to HIF-1α accumulation. HIF-1α hydroxylation on Pro402 and Pro564 and acetylation on Lys532 within the ODD domain facilitates binding with pVHL. As a result, HIF-1α is degraded via the ubiquitin-proteasome pathway. By contrast, sumoylation on Lys391 and Lys477 in the ODD domain may increase HIF-1α stability by competing with hydroxylation and acetylation for the pVHL binding.
Overexpression of HIF-1α in the tumor
Immunohistochemical analysis of human tumor biopsy specimens has revealed the dramatic overexpression of HIF-1α in common cancers (63,64). This is a result of intratumoral hypoxia, genetic alteration and the increased HIF-1α transcriptional activity. In addition to hypoxia, HIF accumulation may occurs as a result of genetic alteration such as the loss or the simple decrease of pVHL (31). Other known causes of increased HIF-1α transcriptional activity include the activation of mitogen-activated protein kinase (65) and the insulin-like growth factor 1 pathway (40,42). In any case, the activated HIF pathway triggers biologic events that are intimately associated with aggressive tumor behavior. Thus, HIF-1α overexpression may be a marker of highly aggressive disease behavior for several different tumor types (Table 1). HIF-1α expression in tumor specimens has been analyzed by immunohistochemistry for cancers of the brain (oligodendroglioma), breast, cervix, melanoma, oropharynx, ovary pancreas, prostate and uterus (endometrial).
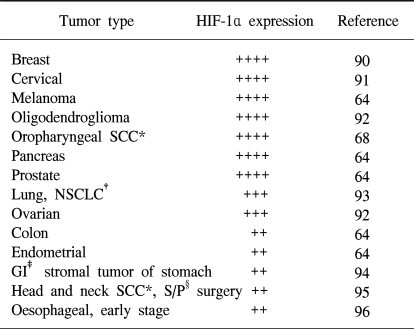
Increased HIF-1α levels in human cancers
HIF-1 targeted therapies
Because tumor cell invasion and migration are reinforced by hypoxic stimuli, hypoxia is a major obstacle for tumor radiotherapy, and it is often a problem in chemotherapy too. HIF-1α-expressing tumors are expected to be resistant to radiation therapy because these kinds of factors denote tumor hypoxia and an enhanced transcription of proteins that will favor tumor cell survival (96). Thus, blocking of HIF-1α activity may be advantageous for inhibiting cancer progression as this would help starve the growing tumors of their oxygen and nutrient supply (66). Recent studies have provided evidence indicating that HIF-1α mediates resistance to chemotherapy and radiation (67,68). Inhibition of HIF-1α activity could, therefore, represent an important component of a combination anti-angiogenic therapy, and strategies for blocking HIF-1α itself or the HIF-1α interacting proteins are under development. HIF-1α antisense therapy might act synergistically with the appropriate immunotherapy. In vivo delivery of antisense to HIF-1α alone by a direct intratumor injection was shown to inhibit tumor growth, but a combination of the two treatments caused marked tumor regression and a sustained antitumor immune response (69). A gene-therapy strategy to block the interaction between HIF-1α and its transcriptional co-activator CBP/p300 led to the attenuation of hypoxia-inducible gene expression and the inhibition of tumor growth in a mouse xenograft model (70). In addition, HIF-1α interacts with the chaperone HSP90, and the HSP90 inhibitor 17-allyl-aminogel-danamycin (17-AAG) induces HIF-1α degradation in a VHL-independent manner (71~73). The small molecule YC-1 (3-(-5'-hydroxy-methyl-2'-furyl)-1-benzylindazole) was also shown to reduce both the HIF-1α levels and xenograft growth (74). The mechanism by which YC-1 reduces HIF-1α levels has not been established, although YC-1 is known to stimulate soluble guanylate-cyclase activity, yet this effect is not required for inhibiting HIF-1α levels. Disruption of microtubule polymerization by 2-methoxyestradiol (2ME2) has also been shown to result in decreased HIF-1α levels (Table 2) (75).
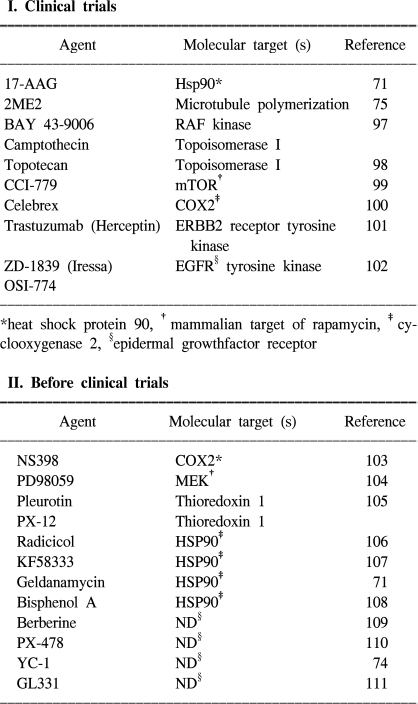
Therapeutic candidates that inhibit HIF-1 activity
Hypoxia response elements (HREs) that are linked to marker genes or prodrug activation systems can be used to selectively activate therapeutics in hypoxic regions (76,77). Gene-therapy vectors that carry pro-apoptotic or anti-proliferation genes driven by HREs can be selectively targeted to cancer cells in hypoxic regions of the tumor (31). For example, in vivo, HRE-mediated trans gene expression was localized adjacent to areas of pyknotic cells and necrosis (76). In addition to the anti-angiogenesis agents, it is clear that many novel therapeutic agents targeting signal-transduction pathways have anti-angiogenic effects. This effect seems to be due in part to the fact that inhibition of the signal-transduction pathways results in decreased levels of HIF-1α (3).
CONCLUSIONS
Hypoxia is a common physiological feature of all tumors, and HIF-1α is a master regulator among a lot of different transcription factors and functions that depend on oxygen tension. Therefore, HIF-1α stabilization is critical for those events that are mediated by hypoxia and dominated by post-translational modification.
Hydroxylation and acetylation are essential to the regulation of HIF-1α protein stability. Under normoxic conditions, the HIF-1α ODD domain encompasses several sequences that mediate O2-dependent ubiquitination of HIF-1α protein through an interaction with pVHL, which is an E3 ubiquitin-protein ligase that targets HIF-1α for proteasomal degradation (58,78~81). Furthermore, ubiquitination of HIF-1α is mediated by an interaction with p53, and this promotes Mdm-2-mediated ubiquitination and proteasomal degradation of HIF-1α through direct interaction with HIF-1α during hypoxia (82,83). Under hypoxia conditions, HIF-1α is stabilized, which is determined by a balance between negative regulators such as p53 and positive unknown regulatory factors, and HIF-1α is then accumulated in the nucleus (84). Stabilized HIF-1α exerts its transcriptional activity by binding to the p300/CBP, the SRC (steroid receptor coactivator-1) family coactivators, nuclear redox regulator Ref-1 and the molecular chaperon heat shock protein 90 (HSP 90) (84~87). They all synergistically enhance HIF-1α-mediated transcriptional regulation under hypoxic conditions. The modulation of HIF-1α stability and its activation requires the interaction of these multiproteins with HIF-1α (88,113,114).
We recently reported that sumoylation increased HIF-1α stability (62). Sumoylation on Lys391 and Lys477 in the ODD domain may increase HIF-1α stability by competing with hydroxylation and acetylation for the pVHL binding.
In addition to angiogenesis, HIF-1α is also a master transcription factor related to cell proliferation and survival, glucose metabolism and iron metabolism. Therefore, HIF-1α is a major player in of the many diseases that generate a hypoxic microenvironment such as cancer, stroke, and heart disease (89). Especially, hypoxic tumor conditions might activate the expression of genes that promote tumor growth, and this would lead to a more aggressive tumor phenotype (43). Thus, the activation of HIF-1α has been associated with a variety of tumors and oncogenic pathways. On the contrary, the blocking of HIF-1α itself or the blocking of HIF-1α interacting proteins inhibits tumor growth (3). HIF-1α antisense therapy is a gene-therapy strategy to block the interaction between HIF-1α and its transcriptional co-activator CBP/p300 and other small molecules such as 17AAG and YC-1, and it has displayed a good possibility as a cancer therapy (69,70). Based on these findings, HIF-1 can be a prime target for anticancer therapies (3,43), and therefore, increased understanding of HIF-1α regulation will provide the cornerstone for novel therapeutic approaches.
Notes
This work was supported by the Creative Research Initiatives Program of the Ministry of Science and Technology, the 21C Frontier R&D Program (FG04-21-01), the Ministry of Science and Technology, and the Korea Health 21 R&D Project (HMP-01-PJ1-PG1-01CH04-0005), Korea.