Characterization of RhoA-mediated Chemoresistance in Gastric Cancer Cells
Article information
Abstract
Purpose
RhoA is a critical transducer of extracellular signals, which leads to organization of actin cytoskeleton, motility, adhesion and gene regulation. The present study aimed to explore whether RhoA influences the susceptibility of gastric cancer cells to chemotherapeutic drugs.
Materials and Methods
SNU638 cells were transfected with a mock vector (pcDNA3.1), RhoA (pcDNA/RhoA), or constitutively active RhoA (pcDNA/caRhoA). MTT assay and Western blot analysis were performed to study the growth response to several chemotherapeutic drugs in the gastric cancer cell line, SNU638, with different RhoA levels.
Results
RhoA significantly enhanced the resistance to lovastatin, 5-FU, taxol and vincristine, but did not affect the sensitivity to cisplatin or etoposide in SNU638. In the Western blot analysis, RhoA decreased the PARP cleavage, which was accompanied by a concurrent reductionin cell death. The gene expression profile after a cDNA microarray analysis demonstrated that RhoA was associated with the differential expression of 19 genes, including those involved in anti-oxidant defense, glucose metabolism, anti-apoptosis and protein turnover.
Conclusion
Gastric cancer cells with a high expression of RhoA could be resistant to chemotherapeutic drugs, such as taxol or vincristine, implying that treatment strategies aimed at inactivation of RhoA might be promising for improving the efficacy of these chemotherapeutic drugs.
INTRODUCTION
As a member of the Ras GTPase superfamily (1), RhoA is an oncogenic and critical component of signaling pathways leading to downstream gene regulation (2~5). RhoA is frequently overexpressed in human cancer (6), but the genetic alteration remains to be reported (7). In terms of function, substantial evidence supports a role for RhoA in adhesion and cytoskeletal reorganization, whereas published data on its participation in cell survival are relatively limited (8,9). Since resistance to chemotherapeutic drugs is one of the major obstacles in the treatment of cancer, whether RhoA expression influences cellular responses to chemotherapeutic drugs in gastric cancer cells was investigated.
In chemotherapy, apoptosis is the predominant mechanism by which cancer cells die. However, even when the apoptotic machinery remains intact, survival signaling may antagonize the cell death by signals, such as growth factor, steroid hormone, neuropeptide and the activation of phosphatidylinositol 3-kinase and Akt (10,11). In view of recent findings, where specific patterns of resistance to chemotherapy can occur depending on the genetic or epigenetic abnormalities of the cancer cells (12~17), this work demonstrated the resistance of gastric cancer cells to cell death in the context of RhoA overexpression. A cDNAmicroarray analysis was then exploited to identify the genes that were differentially expressed according to the RhoA expression. Western blot analysis also showed that the expressions of several chemoresistance-related genes were modulated by RhoA. This is the first report indicating that the increased expression of RhoA in gastric cancer cells may account for taxol or vincristine resistance.
MATERIALS AND METHODS
1) Materials
The lovastatin was obtained from Merck & Co. (San Diego, CA). The other chemicals, protease inhibitors and chemotherapeutic drugs were purchased from Sigma Chemical Co. (St Louis, MO). Fetal bovine serum and tissue-culture media were purchased from Life Technologies, Inc. (Gaithersburg, MA). The enhanced chemiluminescence detection reagents were obtained from Amersham Corp. (Arlington Heights, IL) and the antibodies for RhoA and PARP from Santa Cruz Biotechnology (Santa Cruz, CA).
2) Cell-growth and cytotoxicity assays
The SNU638 human carcinoma cell line (Korean Type Culture Collection, Seoul, Korea) was maintained in RPMI 1,640 (Life Technologies, Gaithersburg, MD), containing 1 mM glutamate, 100 U/ml penicillin, 100 ng/ml streptomycin and 10% FCS. Cells were cultured and passaged at 37℃ in a humidified atmosphere of 5% CO2. For the MTT assay, cells were seeded at 1~5×103 cells/well in a flat-bottomed 96-well tissue-culture plate, treated with or without drugs, and incubated for various times. Before the end of the experiment, the MTT labeling mixture was added and the cells incubated for another 4 h. The viable cell mass was assessed as recommended in the manufacturer's protocol (Promega, Madison, WI). The absorbance of the product was measured at a wavelength of 540 nm, with a reference wavelength of 650 nm. The cell viability was expressed as the ratio of viable cell mass following a given treatment to that of the untreated cells ×100(%).
3) Transfection of SNU638 cells
SNU638 cells were transfected using FuGene (Roche Molecular Biochemicals, Indianapolis, IN), as recommended by the manufacturer, with either a RhoA plasmid in which the full-length RhoA cDNA was cloned into the EcoRI site of the expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) or a similar plasmid expressing caRhoA as the result of the V14 mutation. After transfection, the cells were incubated for 48 h; the medium was changed, and the transfected cells selected for 2 weeks in the presence of geneticin.
4) Western blot analysis
After experimental treatments, the cells were harvested and the cell pellets washed with ice-cold phosphate-buffered saline and lysed in lysis buffer, containing 10 mM Tris (pH 7.5), 150 mM NaCl, 10 mM ethylene diamine tetra acetic acid (EDTA), 1% sodium dodecyl sulfate (SDS), 1 mM sodium orthovanadate and a mixture of protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 µg/ml pepstatin A, 2 µg/ml aprotinin). Lysates were sonicated for 10 s, centrifuged for 20 min at 20,000×g, and stored at -70℃. Equal amounts of lysate proteins were run on 10% SDS-polyacrylamide gels and transferred electrophoretically to nitrocellulose. The nitrocellulose blots were blocked with 5% skimmed milk in Tris-buffered saline with 0.05% Tween-20 (TBST), then incubated at 4℃ with antibody in TBST, containing 5% bovine serum albumin, overnight. Immunoreactivity was detected using enhanced chemiluminescence.
5) cDNA microarray analysis
Total RNA from SNU638/pcDNA3 and SNU638/RhoA cells was purified using the RNeasy mini kit (Qiagen, GmbH, Germany). The relative abundance of a specific transcript was evaluated using the MICROMAX human cDNA microarray system (PE Applied Biosystems, Foster, CA), which contains probes for the detection of approximately 2,400 human gene sequences. A list of a set of genes on the filter is shown in the web site: (http://www.perkinelmer.com/lifesciences). Microarray probe and hybridization were performed as described in the instruction manual. The gene expression images were quantified by measuring the intensity of the signals using Imagene (BioDiscovery, Los Angels, CA). The signal intensity of the filters was compared using the Genespring program (Silicon Genetics, Redwoods, CA).
The fluorescence intensity measured for each sample's chip was normalized to the average fluorescence of the entire sample. To identify differentially expressed RNA species, mocktransfected SNU638/pcDNA3 was considered the baseline sample, with SNU638/RhoA as the experimental sample. Only genes significantly detected in two samples (considered significant based on a p value of <0.05) were considered in the identification of the differentially expressed transcripts. Out of these transcripts, a pair-wise comparison analysis was performed, and the changes denoted as an increase or decrease in the gene expression. The fold change in the signal intensity of a gene was calculated, and only those with more than a 2-fold increase in the RhoA transfectant over that of the base line control were considered as examples of increased gene expression.
6) Statistical analysis
Where appropriate, data were expressed as the mean±SD based on at least three experiments. The Student's t test was used for directly comparing data, with a p value <0.05 considered significant.
RESULTS
1) RhoA expression enhances lovastatin resistance in gastric cancer cells
To study the chemoresistance-associated role of RhoA, stable wild-type RhoA (SNU638/RhoA) and constitutively active RhoA (SNU638/caRhoA) expressing systems in SNU638 gastric cancer cells were generated. Ectopic expression of transfected RhoA or caRhoA was confirmed by Western blot analysis (Fig. 1A). Because RhoA is a target of lovastatin, the response of SNU638/RhoA and SNU638/caRhoA cells to lovastatin were compared with parental cells to verify the activity of the expressed recombinant RhoA. IC50 curves were generated after treatment of cells with a range of different lovastatin concentrations. Forced expressions of RhoA and caRhoA increased the lovastatin resistance in SNU638 cells, with an overall increase in the IC50 from 7.66±0.55 to 23.55±0.75 and 71.88±3.23 µg/ml, respectively (Fig. 1B).
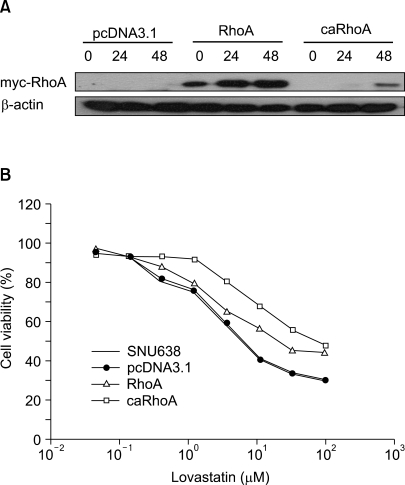
Ectopic expression of RhoA induces resistance to lovastatininduced cell death. (A) Western blot analysis demonstrating ectopic expressions of myc-tagged recombinant RhoA and caRhoA in SNU638 cells. (B) Dose response curves detailing the effect of stable expressions of RhoA and caRhoA on the cytotoxicity of lovastatin in SNU638 cells. Cells (5×103/well) were plated in 96-well plates and treated with lovastatin for 72 h on the following day. A MTT assay was performed to detect live cells. IC50 values were calculated from the sigmoidal dose response curves.
2) RhoA is a differential modulator of chemosensitivity
To examine the role played by RhoA in mediating resistance to chemotherapeutic drugs, dose response curves were generated for the SNU638/pcDNA3.1, SNU638/RhoA and SNU638/caRhoA cells after treatment with the DNA cross-linking agents cisplatin, anti-metabolite 5-fluorouracil, topoisomerase II poison etoposide, anti-microtubule agent taxol and vinca alkaloid vincristine. The cells were plated at equal densities and exposed to increasing doses of each drug for 96 hours, after which time the cell survival was estimated using the MTT assay. SNU638/caRhoA cells displayed a 1.3-fold increase in resistance to 5-FU, with the IC50 value increasing from 0.33±0.03 µg/ml in the SNU638 cells to 0.44±0.03 µg/ml in the SNU638/caRhoA cells (p=0.0094, Table 1). Similar to that observed for 5-FU, the SNU638/caRhoA cells displayed a significant increase in resistance to taxol compared to the SNU638/RhoA cells. Specifically, the SNU638 cells displayed an IC50 value of 4.73±0.26 ng/ml compared with respective IC50 values of 39.12±3.45 and 12.82±0.43 ng/ml in the SNU638/caRhoA and SNU638/RhoA cells (p<0.005), suggestive of RhoA-induced resistance to taxol in these cells (Table 1). SNU638/RhoA and SNU638/caRhoA cells displayed significant increases in resistance to vincristine compared with SNU638/pcDNA cells, displaying IC50 values of 0.47±0.02 and 1.01±0.13 ng/ml, respectively, compared with that of 0.39±0.02 ng/ml in the control SNU638/pcDNA cells (p<0.0005), suggesting RhoA induces resistance to vincristine in these cells (Table 1).
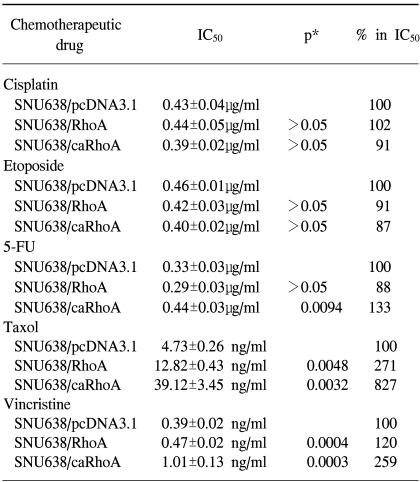
Differential chemosensitivity of SNU638 clones expressing RhoA or caRhoA
However, treatment with cisplatin or etoposide resulted in respective IC50 values of 0.43±0.04 and 0.46±0.01 µg/ml in the parental control cells, whereas the respective IC50 values were 0.44±0.05 and 0.42±0.03 µg/ml in the SNU638/RhoA cells (p>0.05, Table 1). These results reflect a differential resistance pattern of RhoA, which was clearly dependent on the chemotherapeutic drugs (Table 1).
3) PARP cleavage is blocked by RhoA
To examine whether the observed RhoA-induced resistance was due to the inhibition of an apoptotic pathway component, Western blots were conducted and the PARP cleavage after the treatment with drugs analyzed, which induced about 50% apoptosis in the treated cells after 2 days at the given concentration. The PARP assays demonstrated cleavage of the PARP protein when SNU638 cells were treated with cisplatin, etoposide, 5-Fu, taxol and vincristine (Fig. 2). However, in agreement with the cell survival data, SNU638/RhoA and SNU638/caRhoA cells were resistant to the cleavage induced by taxol or vincristine; whereas, the parental or mock cells displayed the characteristic 85 kDa PARP cleavage product.
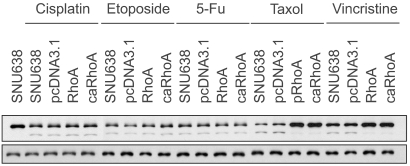
RhoA is a selective modulator of chemotherapeutic drug-induced apoptosis. PARP cleavage assay demonstrates the absence of the cleaved PARP fragment in RhoA (or caRhoA)-transfected cells compared with the parental (SNU638) or mock-transfected cells (pcDNA3.1), after treatments with either 2 µg/ml cisplatin, 5 µg/ml etoposide, 1 µg/ml 5-FU, 2 ng/ml taxol or 10 ng/ml vincristine. Identical blots were reprobed with β-actin as a control for loading.
4) Microarray analysis reveals RhoA targets that may account for drug resistance
The ability of the transcription factor RhoA to increase chemoresistance in a gastric cancer cell line was examined via up-regulation of the target genes. To identify potential target genes, a microarray analysis was carried out, and the differential gene expressions compared between mock and SNU638/RhoA cells. A total of 19 up-regulated genes were identified with at least 2-fold increases in their expressions in the SNU638/RhoA cells (Table 2). Among these there were proteins involved in antioxidant defense and potential detoxification of chemotherapeutic drugs, such as superoxide dismutase. A particular gene, involved in protein ubiquitination, was also up-regulated in the RhoA-transfectants, including deubiquitinating enzyme UnpEL. Genes associated with glucose uptake, such as glucose transporter-like protein-III, were elevated in RhoA-transfected cells. Interestingly, IL-1 receptor-associated kinase (IRAK), which is known to be involved in the IKK phosphorylation and subsequent activation of NF-κB, is also induced by RhoA, implicating that cell death resistance of RhoA could be generated by elevation of these anti-apoptotic proteins.
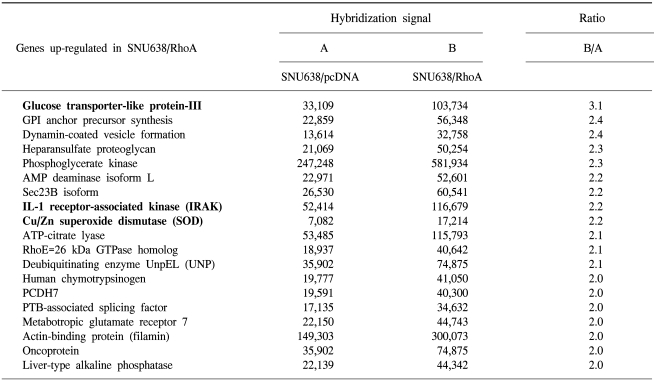
Results of microarray analysis
5) RhoA increases the expression of drug resistance-related proteins
To further examine whether drug resistance-related proteins could be modulated by RhoA, Western blot analyses were performed using SNU638, SNU638/pcDNA3, SNU638/RhoA and SNU638/caRhoA cells. Drug resistance-related proteins, such as vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR), lung resistance protein (LRP) and glutathione S-transferase (GST-π) (18~21), were modulated by either RhoA or caRhoA (Fig. 3), indicating that RhoA expression or signaling induces these proteins, which may subsequently result in cellular chemoresistance.
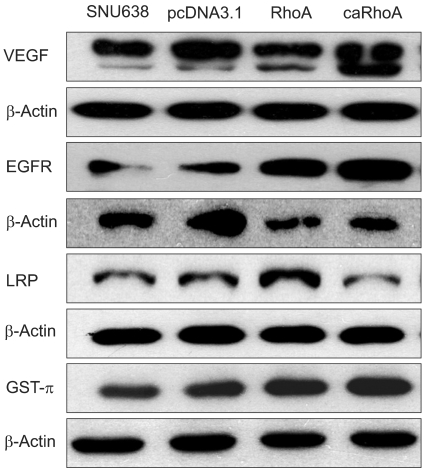
Western blot analyses of chemoresistance-related proteins in total cell lysates from parental (SNU638), mock-transfected (pcDNA3.1), RhoA overexpressing (RhoA) and caRhoA expressing (caRhoA) cells. Identical blots were reprobed with β-actin as a control for loading.
DISCUSSION
Chemotherapeutic drugs induce apoptosis by affecting cell-death pathways. Thus, several ongoing clinical trials are currently under investigation to overcome drug resistance due to modulation of apoptosis (22). In the current study, RhoA was identified as a modulator of taxol and vincristine susceptibility in SNU638 gastric cancer cells, and a possible target of RhoA described, which may account for the resistance. RhoA has been implicated in cytoskeletal reorganization. However, to our knowledge, this is the first report indicating that the expression of RhoA in gastric carcinomas may play a part in cellular resistance to cell death. Our data indicate that an increased expression of RhoA in a gastric cancer cell line induces resistance to taxol and vincristine. Thus, the expression of RhoA may denote a poor prognosis due to the high probability to therapy resistance.
The exact mechanism whereby RhoA modulates cell death remains to be defined, but may reflect an indirect effect of RhoA. Whatever the mechanisms underlying RhoA-mediated inhibition of apoptosis, it is intriguing to note that RhoA-mediated inhibition of cell death is differential depending on the anticancer agents treated. To identify putative targets of RhoA that may account for the resistance to chemotherapy, a cDNA microarray analysis was performed on the RNA extracted from parental and RhoA-transfected cells. The array identified many transcriptional alterations, most with no obvious connection to chemoresistance. However, proteins involved in antioxidant defense and NF-κB signaling were recognized. Specifically, the expressions of superoxide dismutase genes were correlated with RhoA expression. Considering the reports were chemotherapeutic drugs were found to produce a superoxide radical in exerting their cytotoxic effect in cancer cells, the up-regulation of antioxidative protein, such as superoxide dismutase, may alleviate the cytotoxicity. Interleukin-1 receptor-associated kinase, IRAK, was also identified as a target in the RhoA transfected cells. IRAK activates NF-κB by phosphorylation of IKK, which results in the inactivation of IkB. Therefore, over-expressed RhoA may result in activation of NF-κB, and the subsequent up-regulation of anti-apoptotic proteins, such as IAPs or TRAFs (23). It has been suggested that NF-(B may function as an anti-apoptotic survival protein, based on the observation that the inactivation of NF-κB induced apoptosis in several cancer cells (23,24). Therefore, the up-regulation of anti-apoptotic proteins through the activation of NF-κB would be a mechanism for inducing chemoresistance in cancer cells overexpressing RhoA, although this remains to be demonstrated.
CONCLUSIONS
Our study has shown that RhoA signaling activates the survival pathway by inactivating the apoptosis involving cleavage of PARP, which influences the response to cell death in gastric cancer cells. Therefore, gastric cancer cells expressing high levels of RhoA would be resistant to chemotherapeutic drugs, such as taxol or vincristine, implying that treatment strategies aimed at inactivation of RhoA might be promising for improving the efficacy of these chemotherapeutic drugs.
Notes
This work was supported by a grant (KRF-2002-003-E00023) from Korea Research Foundation.