Vps34 Inhibits Hepatocellular Carcinoma Invasion by Regulating Endosome-Lysosome Trafficking via Rab7-RILP and Rab11
Article information
Abstract
Purpose
The role of vacuolar protein sorting 34 (Vps34), an indispensable protein required for cell vesicular trafficking, in the biological behavior of hepatocellular carcinoma (HCC) has yet to be studied.
Materials and Methods
In the present study, the expression of Vps34 in HCC and the effect of Vps34 on HCC cell invasion was detected both in vivo and in vitro. Furthermore, by modulating the RILP and Rab11, which regulate juxtanuclear lysosome aggregation and recycling endosome respectively, the underlying mechanism was investigated.
Results
Vps34 was significantly decreased in HCC and negatively correlated with the HCC invasiveness both in vivo and in vitro. Moreover, Vps34 could promote lysosomal juxtanuclear accumulation, reduce the invasive ability of HCC cells via the Rab7-RILP pathway. In addition, the deficiency of Vps34 in HCC cells affected the endosome-lysosome system, resulting in enhanced Rab11 mediated endocytic recycling of cell surface receptor and increased invasion of HCC cells.
Conclusion
Our study reveals that Vps34 acts as an invasion suppressor in HCC cells, and more importantly, the endosome-lysosome trafficking regulated by Vps34 has the potential to become a target pathway in HCC treatment.
Introduction
Vacuolar protein sorting 34 (Vps34), also known as phosphatidylinositol 3 kinase class III (PI3KC3), is widely distributed in eukaryotic cells. Phosphatidylinositol is the only substrate that can be used by Vps34 to produce phosphatidylinositol 3-phosphate, which is an important messenger located on the cell membrane and endoplasmic reticulum [1].
The first known function of Vps34 was regulating vesicular trafficking in the endosome-lysosome system [1]. Meanwhile, Vps34 has also been implicated in the regulation of autophagy and mammalian target of rapamycin complex 1 (mTORC1) nutrient sensing pathway [2]. Lysosome is the center of endosome-lysosome system and also the platform for mTORC1 signaling and regulation [3]. Therefore, lysosome is not only a substrate recycling center, but also a hub of signal transduction such as nutrient sensing, metabolic adaptation, and quality control of proteins and organelles [4]. As another important component of endosome-lysosome system, endosomes selectively package cell-surface proteins, such as cytokine receptors and adhesion components, and a series of sorting processes determine the fate of internalized proteins, either degradation in lysosomes or recycling back to the plasma membrane [5]. Studies have shown abnormal distribution, quantity, size, and activity of the endosome-lysosome system could affect the cell homeostasis, such as the degradation and inactivation of pathway molecules, which can be commonly observed in cancer cells [6].
Vps34 is a component of Vps34-Beclin1 complex and binds to the central coiled-coil domain of Beclin1, which acts as a platform by connecting Vps34 with different regulatory proteins [7]. Beclin1 has been recognized as a tumor suppressor gene and significantly decreased or lost in many cancer cells, including 75% of ovarian, 50% of breast, and 40% of prostate cancers [8]. However, as a key partner of Beclin1, the role of Vps34 in the onset and progress of cancers is still unclear.
Due to extremely high rate of invasion, metastasis and recurrence of hepatocellular carcinoma (HCC), the five-year survival rate of patients with HCC is only about 5%. The mortality rate of HCC ranks second in China [9]. Cell invasion is the initial step of metastasis [10]. Therefore, exploring the mechanism of invasion is the key to improve the survival rate of HCC patients. However, only a few studies mentioned the relationship between Vps34 and cancers, and the role of Vps34 in HCC remains uncharacterized.
In the present study, we examined change of Vps34 level in HCC both in vivo and in vitro, and more importantly, explored how Vps34 regulated the endosome-lysosome transportation in HCC cells thus to affect the malignant behavior of HCC cells, by modulating Vps34 and related regulatory proteins in endosome-lysosome trafficking.
Materials and Methods
1. HCC tissue samples
Tumor tissue samples were collected from 73 Chinese HCC patients, who underwent curative liver resection for primary tumors between 2014 and 2017 at Shanghai Huashan Hospital. Additional details about these patients’ cohorts are listed in S1 Table.
2. Animals
Nude mice (Slark Laboratory Animal Co., Shanghai, China) were injected once with Vps34 stable transfection SMMC-7721 cells labeled with enhanced-GFP (1×107/mouse) via tail vein. At the 6th week after injection, mice were fasted overnight (12 hours), the enhanced-GFP signal intensity of each nude mouse was measured using a small animal live in vivo optical imaging system (IVIS Lumina K Series III, PerkinElmer, Waltham, MA). Then, mice were sacrificed and livers were processed to make paraffin embedded blocks.
3. Cell culture
Human HCC cell lines were grown in high glucose Dulbecco’s modified Eagle’s medium (Biological Industries, Haemek, Israel) supplemented with 10% fetal bovine serum (Biological Industries), and 100 U/mL penicillin and streptomycin (HyClone, Logan, UT). The feature and source of the cell lines used in this study are listed as follows: L-02 (Hepatocyte, Liver Cancer Research Institute of Fudan University, Shanghai, China), HepG2, Hep3B (HCC with low invasive ability, Liver Cancer Research Institute of Fudan University), SMMC-7721, Huh7, and Bel-7402 (HCC with high invasive ability, Cell Bank of Chinese Academy of Sciences, Shanghai, China), MHCC97-H and HCC-LM3 (HCC with high invasive ability, Liver Cancer Research Institute of Fudan University).
4. Antibodies and reagents
We used the following antibodies: β-actin mouse mAb, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mouse mAb, β-tubulin mouse mAb, Rab7A rabbit mAb, Rab7–interacting lysosomal protein (RILP) rabbit mAb, CD63 rabbit mAb, goat anti-rabbit IgG horseradish peroxidase (HRP) conjugate mAb, goat anti-mouse IgG HRP conjugate mAb, heavy chain of rabbit IgG mAb, mouse anti-rabbit IgG light chain specific HRP conjugate mAb (Proteintech, Rosemont, IL), Vps34/PI3KC3 rabbit mAb, Rab11 rabbit mAb, Lamp1 rabbit mAb, LC3A/B rabbit mAb, matrix metalloproteinase 2 (MMP-2) rabbit mAb, epidermal growth factor receptor (EGFR) rabbit mAb, p-EGFR (Y1068) rabbit mAb (Cell Signaling Technology, Danvers, MA), Lamp1 mouse mAb, MAPLC3β mouse mAb (Santa Cruz Biotechnology, Dallas, TX), TSG101 rabbit mAb (Abcam, Cambridge, MA), EGFR mouse mAb (Bioss, Woburn, MA), AF594 AffiniPure goat anti-rabbit, AF647 AffiniPure goat anti-mouse, AF594, AffiniPure donkey anti-rabbit, DyLight 488 horse anti-mouse (Vector Laboratories, Burlingame, CA). Reagents we used in this study were listed: Vps34-IN-1 (MedChemExpress, Monmouth Junction, NJ), CQ (Sigma-Aldrich, St. Louis, MO), human epidermal growth factor (EGF; Sigma-Aldrich).
5. Immunohistochemistry staining
Slides from HCC patients were baked overnight and then deparaffinized and rehydrated. Antigen retrieval was performed using citrate buffer (pH 6.1) and microwave treatment. Hydrogen peroxide (3%) was applied at room temperature (RT) for 20 minutes. After blocking by 5% goat serum at RT for 1 hour, primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Secondary antibody incubation was performed for 1 hour at RT. DAB (Agilent, Beijing, China) was applied for staining. Images were taken by a light microscope (Nikon Y-THM, Tokyo, Japan). Scoring of Vps34 was performed by two pathologists following the protocol according to the study by Ma et al. (2010) [11]. The immunostaining positivity (P) was scored as follows: 0, < 5%; 1, 5%–25%; 2, 25%–50%; 3, 50%–75%; and 4, > 75%. The immunostaining intensity (I) was scored as follows: 1+ (weak); 2+ (moderate), or 3+ (intense). The Score of immunohistochemistry staining was represented by Positivity (P)×Intensity (I) and defined as follows: −(0–1); 1+ (2–3), 2+ (4–6), and 3+ (> 6).
6. Immunofluorescence staining
Cells were grown to 40%–60% confluence on 24-well chamber glass slides (JingAn Biological, Shanghai, China). After experimental treatment, cells were fixed in ice-cold phosphate buffered saline (PBS) containing 4% paraformaldehyde (Sigma-Aldrich). Then, for non-membrane targets (such as Lamp1, Rab7, LC3β, and tubulin), cells were permeabilized with 0.05% Triton X-100 (Beyotime, Shanghai, China) for 5 minutes. Then followed by blocking in 1% bovine serum albumin (BSA) for 1 hour. For membrane targets (such as EGFR), cells were blocked with 1% BSA directly after fixation without permeabilization. Primary antibodies were applied overnight at 4°C. Afterwards, the cells were washed with PBS and incubated with secondary antibodies for 2 hours at RT. Subsequently, slides were mounted with fluoroshield mounting medium with DAPI (Abcam). Slides were visualized and photographed by a fluorescence microscope (Zeiss AXIO Imager, Jena, Germany) with the inform software (Zeiss ZEN Lite).
7. Western blot analysis and co-immunoprecipitation
The cells were lysed with ice-cold RIPA lysis buffer (Bimake, Shanghai, China). A BCA Protein Assay Kit (Pierce, CA) was used to determine the protein concentration. About 10–30 μg of total protein was resolved by 8%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF membrane (Millipore, Billerica, MA). The membranes were blocked with 5% non-fat dried milk in 1× TBST buffer and incubated at 4°C for at least 16 hours with the indicated primary antibodies. Secondary antibodies conjugated with HRP anti-rabbit (1:2,000, Proteintech) or anti-mouse (1:2,000, Proteintech) were used. The membranes were incubated with Immobilon Western Chemiluminescent HRP Substrate (Thermo Fisher Scientific, Waltham, MA). The chemiluminescent signal was detected by the ImageQuant LAS 4000 system (GE Healthcare, Chicago, IL). The band intensities were quantified using ImageJ software and normalized to the expression of β-actin or GAPDH (loading control) and expressed as fold-change relative to control group.
For co-immunoprecipitation, cell lysate was incubated with a chosen antibody overnight at 4°C followed by incubation with magnetic beads (Pierce) for 1 hour at RT. Beads were washed twice with lysis/wash buffer and once with purified water. The antigen/antibody complex was eluted. Then, samples were subjected to SDS-PAGE for separation and detection.
8. RNA isolation and quantitative real-time PCR (qRT-PCR)
According to the manufacturer’s protocol, cells were homogenized in Trizol (Invitrogen, Carlsbad, CA) and total RNA was isolated. In addition, cDNA was synthesized using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Tokyo, Japan) according to the manufacturer’s protocol. Quantitative real-time PCR was then performed using SYBR Fast qPCR Mix (TaKaRa). Primers were constructed and purchased from Genewiz, Shanghai, China: Vps34 forward primer (5′ to 3′), GTCTGGCCTAATGTAGAAGCAG; Vps34 reverse primer (5′ to 3′), GGCAAGACGGCTCATCTGAT; β-actin forward primer (5′ to 3′), AGGCACCAGGGCGTGAT; β-actin reverse primer (5′ to 3′), GCCCACATAGGAATCCTTCTGAC.
9. Transfection and lentiviral delivery of shRNA
Plasmids were brought from Genomeditech and Genepharma. The RNAi target sequences are listed: shVps34 (5′ to 3′), ACAGAGUGAGCAGUACCAAdTdT; shRILP (5′ to 3′), GGAGCTTGTGTACCATCTAGC; shRab11 (5′ to 3′), GGCAGTTCCTACAGATGAAGC; shNT (5′ to 3′), GTTCTCCGAACGTGTCACGT.
For transient knockdown of protein expression, plasmid was transfected into cells L-02, SMMC-7721, and HepG2 cells by using TransIT-X2 Transfection Reagent (Mirus Bio, Madison, WI) according to the manufacturer’s protocol. For stable knockdown experiments, lentiviral particles were generated using psPAX2 packaging plasmid and pMD2.G envelope plasmid (Addgene, Watertown, MA) according to manufacturer’s protocol. After infecting lentivirus, the cells were selected by puromycin, blasticidin or G418 for at least a week to stably express the shRNA of interest.
10. Transwell invasion assay
Transwell inserts (Corning, New York, NY) were coated with Matrigel (Corning). Cells (2×104) were plated on top of the inserts in serum-free conditions. Cells were allowed to invade for 24 hours. Then the cells were fixed in 4% paraformaldehyde for 20 minutes and stained with 0.1% crystal violet for 30 minutes. The cells remaining on the top of the insert (those that did not invade) were removed with a cotton swab. Membranes were removed completely from the chamber and mounted with resin and cover slip on the slides. At least six pictures per membrane were taken under 20× field using a light microscope (Nikon Y-THM).
11. Wound healing assay
Cells were seeded into a six-well plate and grown to confluence for more than 90%. The wounding took place with a 200-μL pipette tip cutting longitudinally in the well. Wells were washed and followed by a medium change. Baseline (0-hour), 24-hour, and 48-hour phase contrast images (×5) were taken with an inverted microscope (Leica DMi8, Wetzlar, Germany). The mean scratch areas of at least 16 images of each sample and every time point were measured and analyzed with Image J software (National Institutes of Health, Bethesda, MD). The scratch area covered at 0 hour, 24 hours, or 48 hours of each well was calculated and used to represent the migration ability of each sample.
12. Statistics
GraphPad Software Prism 8.0 (GraphPad Software Inc., San Diego, CA) was utilized to perform all statistical analysis. ANOVA or t tests were performed to indicate statistical significance. All graphs were created using this software package.
Results
1. Decrease of Vps34 in HCC and the correlation between Vps34 with invasion ability
We first analyzed the Vps34 expression in various cancers from the Oncomine database. The number of low expression studies (lower Vps34 mRNA expression in cancer tissue compared to normal tissue) was more than the high expression studies (higher Vps34 mRNA expression in cancer tissue compared to normal tissue) (S2A Fig.). We also listed representative studies of cancer types that only had low expression studies in top 10% gene rank (S2B-S2G Fig.). Furthermore, there were 5 HCC studies in the whole database with a p-value of < 0.05 after excluding the cirrhosis and cholangiocarcinoma studies. Three of them had lower expression of Vps34 in HCC tissues (S3A-S3C Fig.), and two of them were higher expression studies (S3D and S3E Fig.). The meta-analysis showed an overexpression p-value of 0.043 and an under-expression p-value of 1.18×10−5 which indicated that under-expression was much more significant than overexpression (S3F Fig.). We supposed that Vps34 mRNA was downregulated in HCC compared to normal liver.
Next, immunohistochemistry staining was performed on HCC specimens (n=73) containing carcinoma and peri-carcinoma tissues to verify the hints collected from Oncomine database mentioned above. The expression levels of Vps34 were divided into four grades (−, +, ++, and +++) and defined as low or high expression group (Fig. 1A). Compared to peri-carcinoma tissues (79.45% of them had high Vps34 expression, 20.55% had low Vps34 expression), there was a markedly decreased Vps34 expression in the carcinoma tissue (32.88% had high Vps34 expression, 67.12% had low Vps34 expression) (Fig. 1B and C). Then, by analyzing clinical data, we found the correlation between Vps34 level in carcinoma tissue and microvascular invasion (MVI) as indicated in S1 Table. Compared to the patients without MVI, the Vps34 level of carcinoma tissue in cases with invasion was significantly decreased (Fig. 1D).
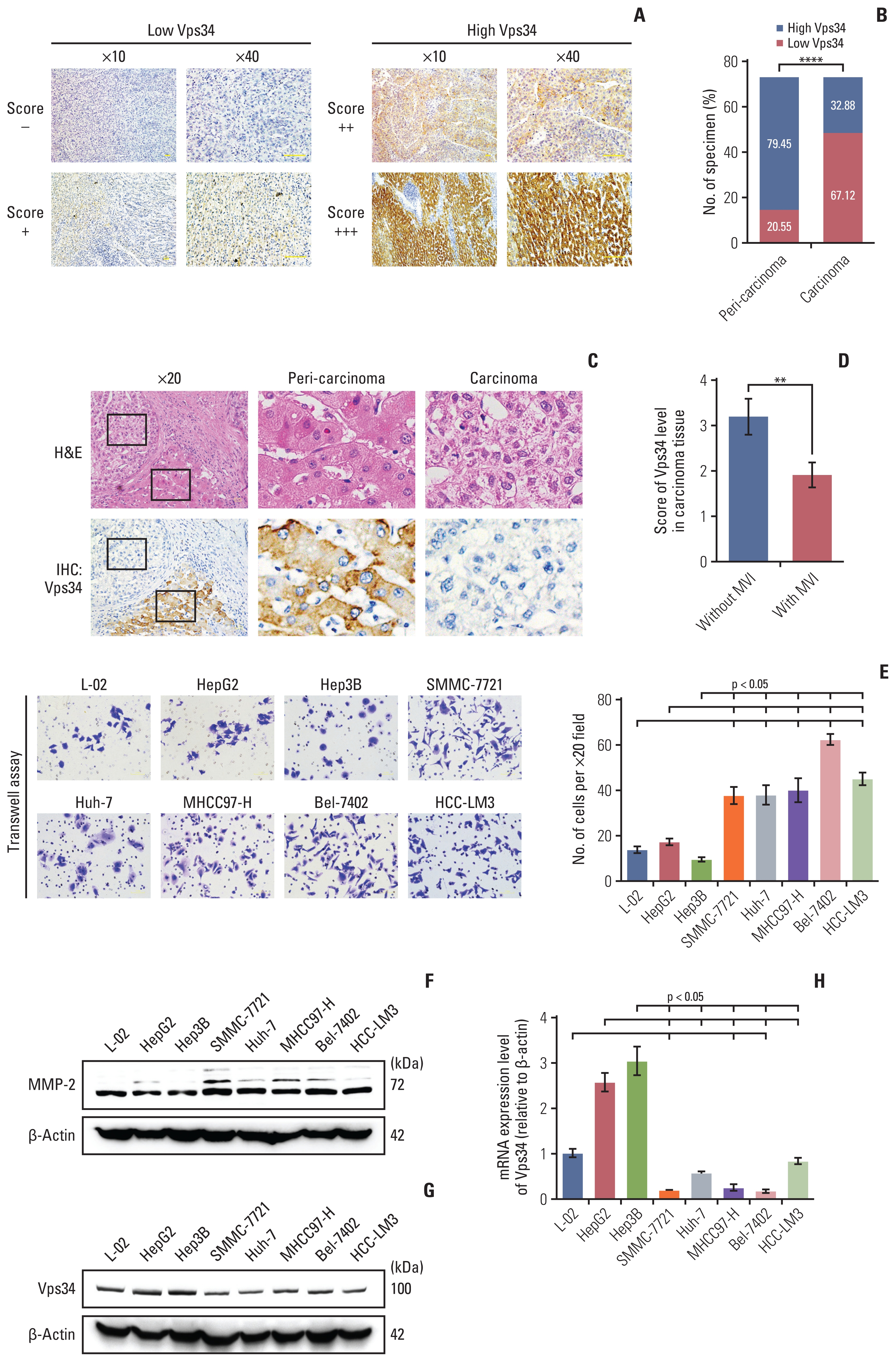
Vacuolar protein sorting 34 (Vps34) was decreased in hepatocellular carcinoma (HCC) and inversely correlated with the invasion ability. (A) Immunochemistry staining of Vps34 in peri-carcinoma tissue or carcinoma tissue of HCC specimens. Scoring mentioned in materials and methods. The expression levels of Vps34 were divided into four grades (−, +, ++, and +++) and defined as low or high expression group. (B) Quantification of high or low Vps34 cases in peri-carcinoma tissue or carcinoma tissue of HCC specimens (n=73), respectively. (C) Representative images of H&E and immunohistochemistry staining of Vps34 in HCC specimens that have both carcinoma tissue and peri-carcinoma tissue. (D) Vps34 scores of carcinoma tissue in patients with or without microvascular invasion (MVI). (E) Transwell assay of the hepatocyte cell line (L-02) and HCC cell lines (HepG2, Hep3B, SMMC-7721, Huh-7, MHCC97-H, Bel-7402, and HCC-LM3). (F) Western blot demonstrated the MMP-2 protein level in above cell lines. (G, H) Protein (G) and mRNA (H) levels of Vps34 in hepatocyte cell line (L-02), HCC cell lines with lower invasive potentials (HepG2 and Hep3B) or HCC cell lines with higher invasive abilities (SMMC-7721, Huh-7, MHCC97-H, Bel-7402, and HCC-LM3). MMP-2, matrix metalloproteinase 2. Data were showed as means±standard error of mean. **p < 0.01, ****p < 0.0001.
We then carried out Transwell assays and examined the MMP-2 level of hepatocytes and seven HCC cell lines to conform their invasive abilities according to Ye et al.’s study [12], and examined the Vps34 level as well (Fig. 1E and H). In SMMC-7721, Huh-7, MHCC97-H, Bel-7402, and HCC-LM3 cell lines, which have higher invasive capabilities (Fig. 1E and F), both protein and mRNA level of Vps34 were significantly decreased compared to HCC cell lines with lower invasive potentials (i.e., HepG2 and Hep3B) and hepatocytes (L-02) (Fig. 1G and 1H). These results above indicated a downregulation of Vps34 in HCC and an inversed correlation between Vps34 level and HCC invasion.
2. Regulation of invasive ability by Vps34 in HCC cells
To investigate whether Vps34 could affect the invasive ability of HCC cells, Vps34 was knocked down by Vps34-shRNA (shVps34) in HepG2 cells, which have a higher Vps34 level and lower invasive ability. After Vps34 knocking down, a morphological change of epithelial-mesenchymal transition (Fig. 2A) was observed, accompanied by decreased epithelial marker E-cadherin, increased mesenchymal marker vimentin, increased MMP-2 (Fig. 2B), enhanced invasive ability (Fig. 2C) and also migration ability (Fig. 2D) compared to control (shNT). We further overexpressed Vps34 in SMMC-7721 cells, which have a lower Vps34 level and higher invasive ability (Fig. 2E). Compared to control (vector), Vps34 overexpression increased E-cadherin, decreased vimentin and MMP-2 (Fig. 2E), and reduced invasion and also migration ability (Fig. 2F and G). Moreover, SMMC-7721 cells with or without Vps34 overexpression (both were enhanced-GFP labeled) were injected into nude mice, there was no significant difference between mice injected with SMMC-7721 cells with Vps34 overexpression and vector control, but the enhanced-GFP signal of the whole body showed numerical decrease in Vps34 overexpression group by using in vivo imaging system (S4A Fig.). Furthermore, by immunohistochemistry staining of GFP, we found there were significantly less GFP-positive cells in liver sections of Vps34 overexpression group compared to vector control (S4B Fig.). These results suggested that Vps34 could regulate the invasive ability of HCC cells.

Vacuolar protein sorting 34 (Vps34) regulated the invasive ability of hepatocellular carcinoma (HCC) cells. (A) H&E staining of HepG2 cells with transfection of shRNA against Vps34 (shVps34) or the non-target shRNA control (shNT). (B) Western blot demonstrated the efficiency of Vps34 knockdown and the protein expression of epithelial marker (E-cadherin), mesenchymal marker (Vimentin) and MMP-2 in HepG2 cells with shVps34 or shNT transfection. (C) Transwell assay of HepG2 cells after shVps34 transfection. (D) Wound healing assay of HepG2 cells expressing shNT or shVps34. (E) Western blot examined the protein level of Vps34, E-cadherin, vimentin, and matrix metalloproteinase 2 (MMP-2) in SMMC-7721 HCC cells expressing vector control (Vector) or exogenous upregulation (Vps34). (F, G) Transwell assay (F) and wound healing assay of SMMC-7721 cells (G) with Vps34 or vector transfection. Data were showed as means±standard error of mean. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3. Role of Vps34 in the lysosome distribution of HCC cells
Because Vps34 plays crucial roles in the context of vesicular trafficking, including endocytosis and also autophagy [13], whether HCC and normal liver cell differ in vesicular trafficking status is of importance. A series of vesicular markers including: LC3β (autophagosome), Rab7 (late endosome), and Lamp1 (lysosome) were examined by immunohistochemistry staining in HCC tissue (S5 Fig.). Both LC3 and Rab7 decreased in carcinoma tissue compared to peri-carcinoma (S5A Fig.). Interestingly, we did not find significant change in Lamp1 expression level, but find obvious change of Lamp1 localization, showing Lamp1 accumulated in the periphery area of the carcinoma cells whereas located in the juxtanuclear region of the cells in the peri-carcinoma tissue (Fig. 3A, S5A Fig.). Moreover, immunofluorescence staining of the above vesicular markers was performed in HCC cells and hepatocytes. Compared with hepatocytes (L-02), which had a juxtanuclear population of Lamp1, HCC cells with higher invasive ability (MHCC-97H and SMMC-7721) showed reduced co-localization of LC3 and Rab7 with Lamp1, and more importantly, lost the phenomenon of juxtanuclear lysosome aggregation (JLA) and showed a dispersed distribution of lysosomes (Fig. 3B, S5B Fig.).
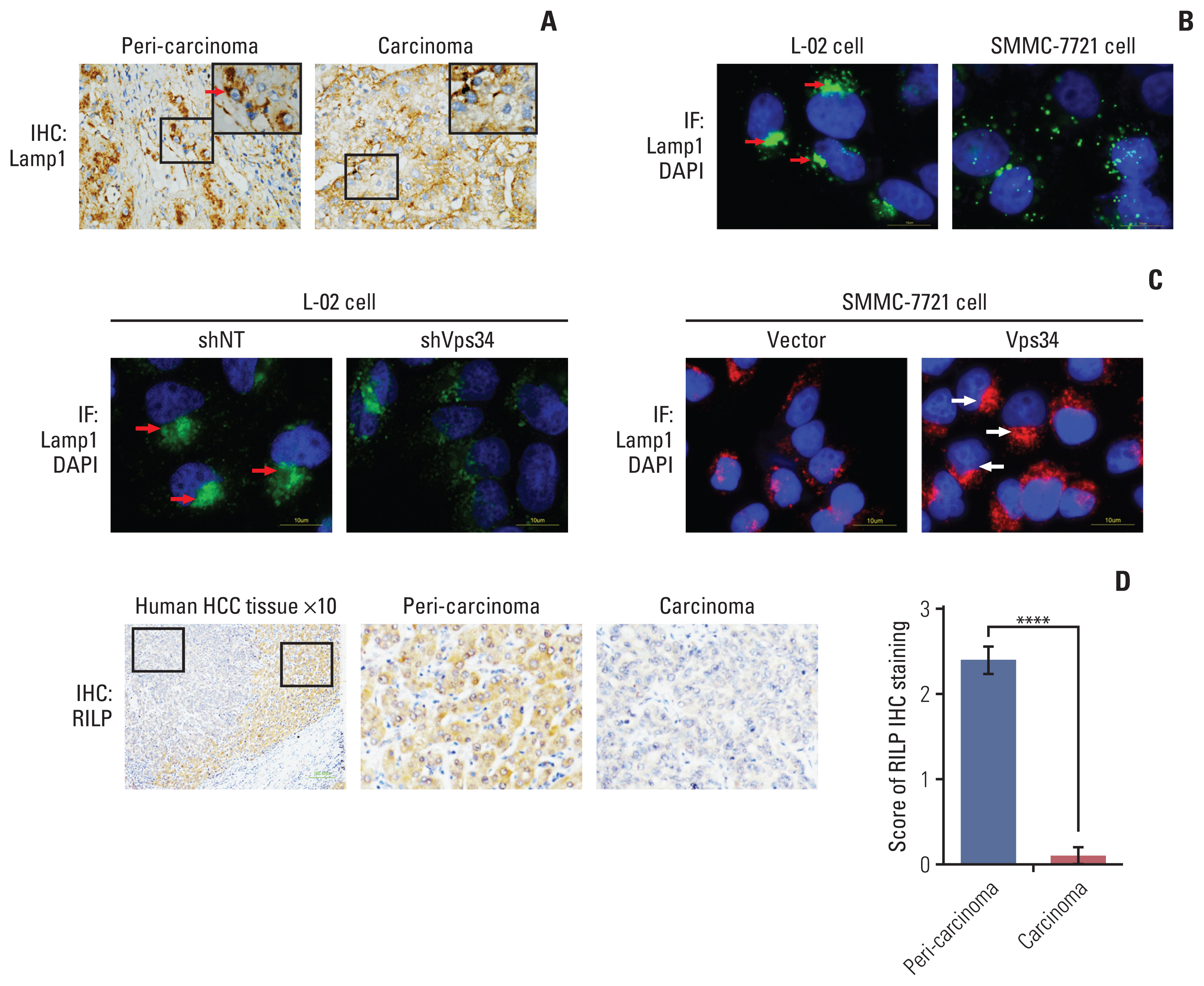
Vacuolar protein sorting 34 (Vps34) regulated the lysosome distribution in hepatocellular carcinoma (HCC) cells. (A) Immunohistochemistry (IHC) staining of Lamp1 (lysosomal marker) in carcinoma and peri-carcinoma tissue showed that juxtanuclear lysosome aggregation (JLA) existed in peri-carcinoma tissue, but not in carcinoma tissue. (B) Immunofluorescence (IF) staining of Lamp1 (green) and DAPI (blue) in normal hepatocyte cell line (L-02) and HCC cell line with high invasive ability (SMMC-7721). Red arrows indicated the JLA. (C) Immunofluorescence staining of Lamp1 showed the lysosome distribution in L-02 cells expressing shNT or shVps34, and in SMMC7721 expressing blank vector or exogenous Vps34. Arrows indicated the juxtanuclear lysosome aggregation. (D) Immunohistochemistry staining of Rab7-interacting lysosomal protein (RILP) in HCC patient specimens containing both carcinoma and peri-carcinoma regions (n=10). Data were showed as means±standard error of mean. ****p < 0.0001.
To investigate the correlation between the Vps34 and JLA of HCC cells, we first knocked down Vps34 in the hepatocytes L-02, and found that the Lamp1 positive puncta changed from juxtanuclear clusters to a dispersed distribution, indicating the disappearance of the JLA. And then we overexpressed Vps34 in SMMC-7721 cells. Compared to the phenomenon showing dispersed distribution of lysosomes in vector control, JLA was observed in SMMC-7721 cells with Vps34 overexpression (Fig. 3C).
In addition, we observed the expression of RILP, an important downstream effector of Rab7 controlling lysosomes moving along the minus-end of the juxtanuclear microtubule organizing center [14], in carcinoma and peri-carcinoma tissues of HCC, and surprisingly found that RILP was significantly decreased in carcinoma compared to peri-carcinoma tissue, which was consistent with the change of Vps34 (Fig. 3D). From the above observation, we deduced that Vps34 could regulate lysosomal trafficking.
4. Inhibition of HCC invasion by Vps34 through the Rab7-RILP mediated lysosomal trafficking
To investigate the detailed mechanism of Vps34 in regulating HCC invasion via lysosomal trafficking, we overexpressed Vps34 in SMMC-7721 cells, and found that compared to vector control, both Rab7 and RILP were markedly increased after Vps34 overexpression, whereas, Lamp1 did not show change in protein level (Fig. 4A). In addition, the interaction of Rab7 with RILP or with Vps34 was also increased in SMMC-7721 cells with Vps34 overexpression (Fig. 4B). Then, we knocked down the RILP in SMMC-7721 cells with or without Vps34 overexpression (S6A Fig.), and found that the JLA induced by Vps34 overexpression in SMMC-7721 cells was significantly reduced after downregulation of RILP (Fig. 4C).
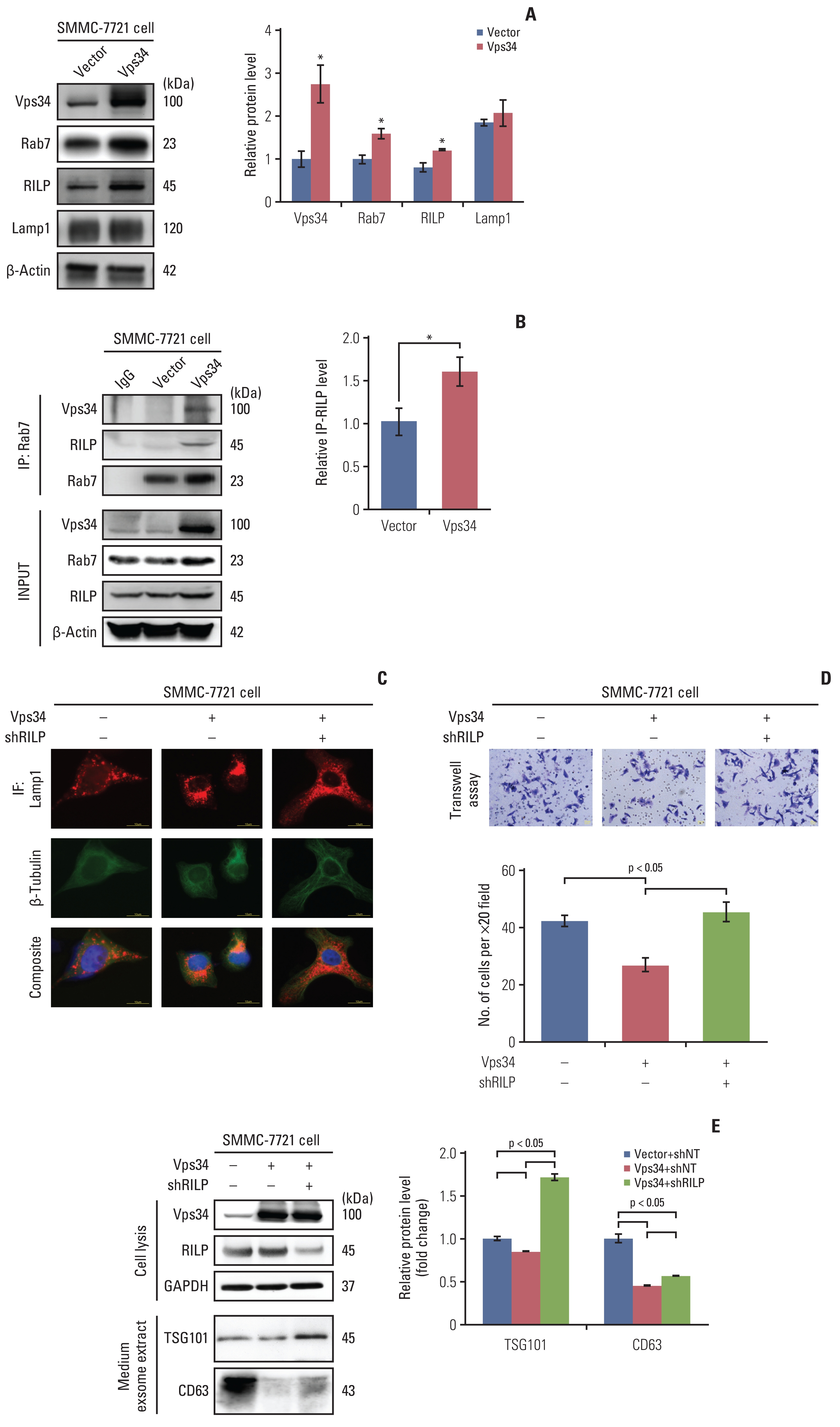
Vacuolar protein sorting 34 (Vps34) inhibited the hepatocellular carcinoma invasion through the Rab7–Rab7-interacting lysosomal protein (RILP) pathway mediated lysosomal trafficking. (A) Western blot examined the protein level of Vps34, Rab7, RILP, and Lamp1 in SMMC-7721 cells with exogenous Vps34 or vector control. (B) Immunoprecipitation of Rab7 and RILP and quantification of RILP pulled down by Rab7 from SMMC-7721 cells with Vps34 overexpression and vector control. Western blot examined the input and immunoprecipitated level of RILP. (C) Double immunofluorescence staining of Lamp1 (red) and β-tubulin (green) in SMMC-7721 cells with or without exogenous Vps34 and/or shRILP transfection. (D, E) Transwell invasion assay (D) and western blots of the TSG101 and CD63 (exosome markers) from the exosome extract (E) of SMMC-7721 cells with or without exogenous Vps34 and/or shRILP transfection. IF, immunofluorescence. Data were showed as means±standard error of mean. *p < 0.05.
With downregulation of RILP and its suppressive effect on JLA, the invasion of SMMC-7721 cells was increased, which inversed the invasive-inhibition effect of Vps34 overexpression on SMMC-7721 cells (Fig. 4D). In addition, two exosome markers, TSG101 and CD63, were downregulated by Vps34 overexpression in the cell culture medium of SMMC-7721 cells, but was markedly up-regulated by RILP knockdown subsequently (Fig. 4E). Therefore, the results above suggested that the inhibition effect of Vps34 on HCC invasion was at least partially mediated by Rab7-RILP regulated lysosomal trafficking.
5. Function of Vps34 as a HCC invasion suppressor by regulating Rab11 mediated endocytic recycling
Then we knocked down Vps34 in HepG2 cells to further investigate its effect on HCC invasion, but surprisingly, rather than the change of lysosomal trafficking, we observed significant macrovesicles in HepG2 cells after Vps34 knockdown (Fig. 5A), which could not be simply explained by the results from Fig. 4. By immunofluorescence staining, we found these vesicles were Lamp1-positive or both Rab7 and Lamp1 positive (Fig. 5B), but neither LC3β positive nor LC3β and Lamp1 co-positive (S6B Fig.). Parallel findings could be observed in cells with a new Vps34 inhibitor (Vps34-IN1) (Fig. 5B). Those large Rab7-positive vesicles was also observed in mouse HCC tissues, accompanied by lower expression of Vps34 (S6C Fig.). These results indicated that endocytosis was involved in this special morphological change showing large vesicles.
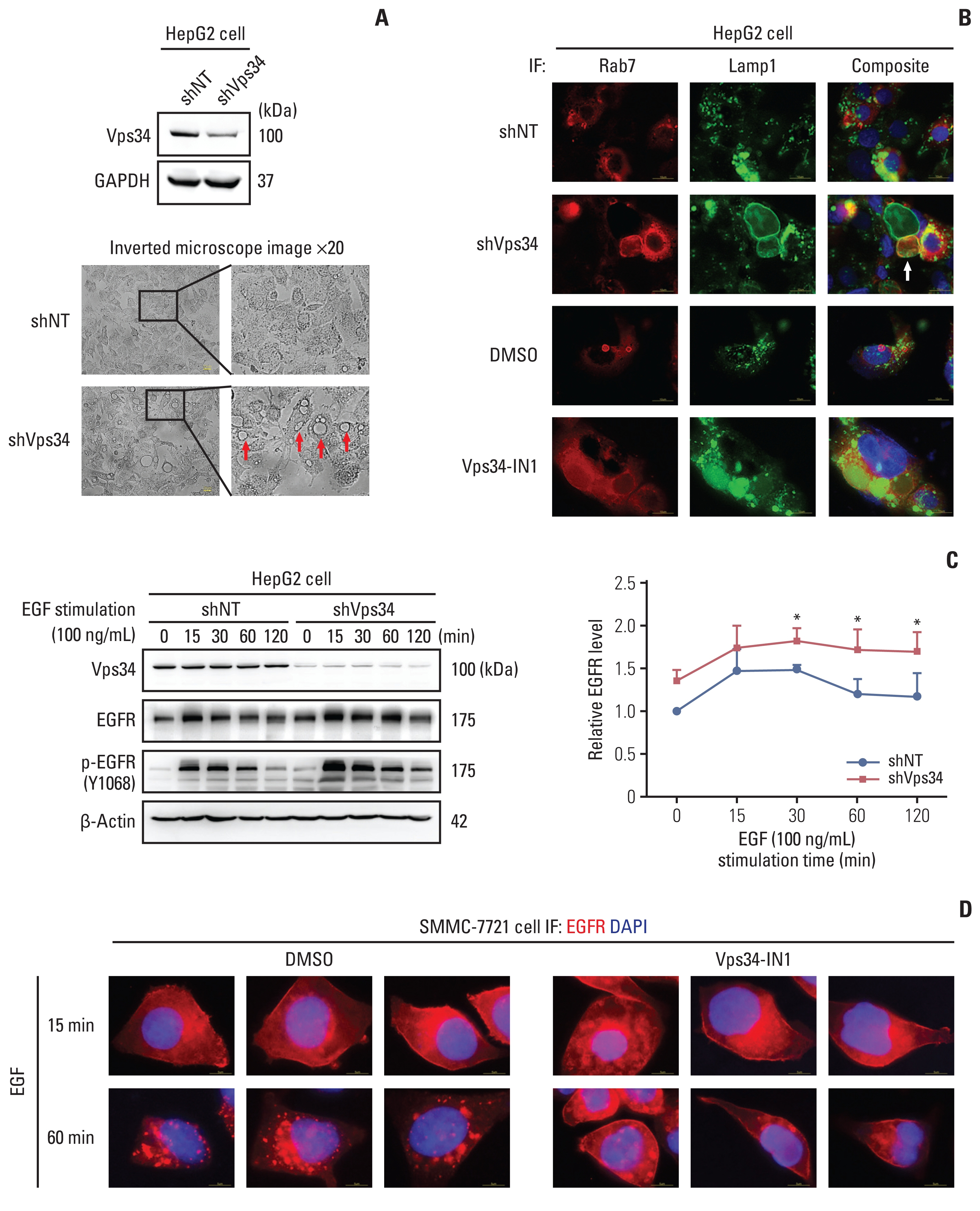
Vacuolar protein sorting 34 (Vps34) deficiency affected the endosome-lysosome system and epidermal growth factor (EGF) induced epidermal growth factor receptor (EGFR) signaling in hepatocellular carcinoma cells. (A) Western blot examined the protein level of Vps34 in HepG2 cells with shVps34 or shNT transfection, and representative images of Vps34 in HepG2 cells with shVps34 or shNT transfection by an inverted microscope. (B) Immunofluorescence (IF) staining of the late endosomal marker Rab7 (red) and Lamp1 (green) in HepG2 cells with shVps34 or shNT transfection, and in HepG2 cells with Vps34-IN1(4 μM, 24 hours) treatment, a Vps34 inhibitor, and DMSO control treatment as well. Arrow indicated the abnormal accumulation of macrovesicles. (C) Western blot examined the protein level of Vps34, EGFR, and p-EGFR (Y1068) in HepG2 cells with shVps34 or shNT transfection after stimulating with EGF (100 ng/mL) for 0, 15, 30, 60, and 120 minutes. (D) Immunofluorescence staining of EGFR (red) in SMMC-7721 cells with Vps34-IN1 or DMSO control treatment, after stimulating with EGF (100 ng/mL) for 15 or 60 minutes. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. Data were showed as mean±standard error of mean. *p < 0.05.
To determine whether autophagy plays role in the effect of Vps34 in regulating HCC invasion, the autophagy flux was observed by examining the content of LC3 dynamic changes with chloroquine (CQ) in HepG2 with or without Vps34 knockdown. After downregulating of Vps34 in HepG2 cells, LC3-II/I level with the presence of CQ did not show significant change (indicating the production of autophagosome), neither did LC3-II (CQ-PBS), which represented the level of autophagosome elimination, despite that p62 showed an increase in Vps34 knockdown cells (S6D Fig.). Therefore, the blockage of endosome-lysosome fusion and degradation rather than autophagosome production and its fusion with lysosome was the predominate outcome when Vps34 was insufficient in HepG2 HCC cells.
Thus, we next focus on endosome-lysosome system in the following study. The canonical regulation mode of impaired endocytosis to cancer invasion and proliferation is achieved through inhibited degradation of overexpressed receptor tyrosine kinases (RTKs) and enhanced recycling, providing sustained signaling [5]. EGFR belongs to RTKs, which are internalized and cleared from cell surface via endosome-lysosome system [15]. To further investigate the role of Vps34 on endosome-lysosome trafficking in HCC cells, EGFR dynamic change was examined with EGF stimulation. As shown in Fig. 5C, Vps34 knockdown enhanced the amount of both EGFR and phospho-EGFR (Y1068), which reached a peak at 15 minutes of EGF treatment and gradually decreased with increasing time from 15 to 120 minutes, compared to control. The expression of EGFR on the cell membrane decreased at 60 minutes of EGF stimulation in control cells, whereas, it remained at a high level in cells with Vps34 inhibition (Fig. 5D).
Meantime, we found Rab11, a marker of recycling endosome, was increased in HepG2 cells with Vps34 knockdown, and decreased in SMMC-7721 cells with Vps34 overexpression (did not change by RILP knockdown) (Fig. 6A and B). We also observed a significant upregulation of Rab11 in carcinoma tissue compared to peri-carcinoma region of HCC specimens (Fig. 6C). When we knocked down Rab11, increased EGFR and p-EGFR level and enhanced invasion caused by Vps34 knockdown could be reversed partially by Rab11 knocking down (Fig. 6D and E). The results above indicated that Vps34 deficiency also promoted the invasion ability of HCC cells by regulating Rab11 mediated RTKs endocytic recycling, besides of its regulation on Rab7-RILP mediated lysosomal trafficking.
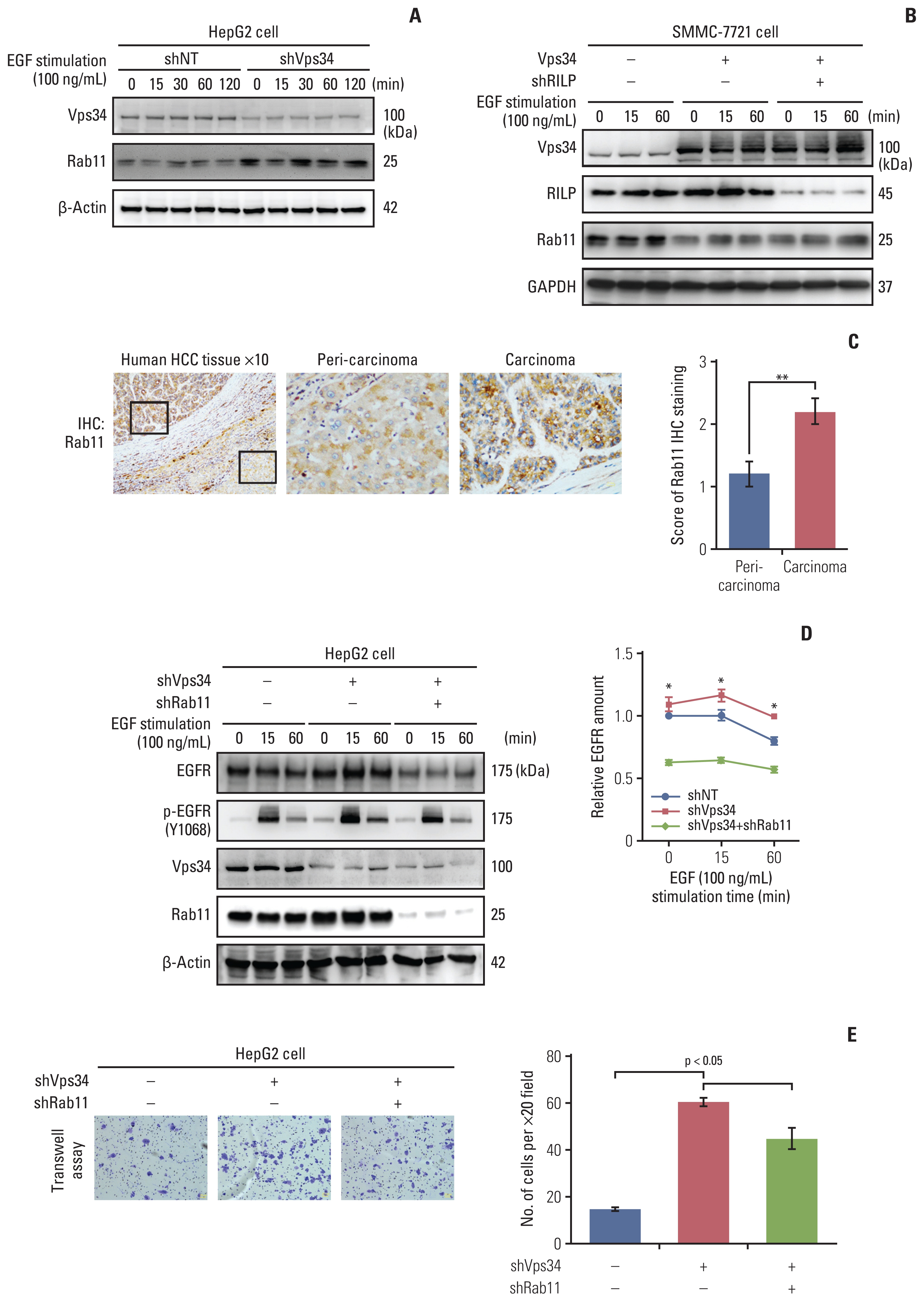
Vacuolar protein sorting 34 (Vps34) affected the invasion ability of hepatocellular carcinoma (HCC) cells by regulating Rab11 mediated endocytic recycling. (A) Western blot examined the protein level of Vps34 and Rab11 in HepG2 cells with shVps34 or shNT transfection after stimulating with epidermal growth factor (EGF) (100 ng/mL) for 0, 15, 30, 60, and 120 minutes. (B) Western blot examined the protein level of Vps34, Rab7-interacting lysosomal protein (RILP) and Rab11 in SMMC-7721 cells with or without exogenous Vps34 and/or shRILP transfection after stimulating with EGF (100 ng/mL) for 0, 15, and 60 minutes. (C) Immunohistochemistry staining of Rab11 in HCC patient specimens containing both carcinoma and peri-carcinoma regions (n=5). (D) Western blot examined the protein level of Vps34, Rab11, EGFR, and p-EGFR (Y1068) in HepG2 cells with or without shVps34 and/or shRab11 transfection after stimulating with EGF (100 ng/mL) for 0, 15, and 60 minutes. (E) Representative images and quantification of Transwell assay in HepG2 cells with or without shVps34 and/or shRab11 transfection. Data were presented as mean±standard error of mean. *p < 0.05, **p < 0.01.
Discussion
Vps34 have been shown to govern vesicular trafficking in many cells and organs [16–18]. However, how Vps34 affects the cancer developing via its important function on substrate transportation-nutrient recycling balance is only beginning to be understood. In the present study, we were the first to elucidate a significant decrease of Vps34 in HCC from various aspects, including database, HCC patient specimens and cultured HCC cells, and an inverse relationship between Vps34 level and the HCC invasiveness. Furthermore, according to the detailed mechanism study, we summarized that there are two mechanisms of Vps34 in inhibiting the HCC invasion: one is Vps34 could promote lysosomal juxtanuclear accumulation through Rab7 and RILP pathway when Vps34 is sufficient, thus to reduce the secretion of exosomes and invasion; the other one is Vps34 deficiency could reduce the endocytic degradation and promote the Rab11 mediated endocytic recycling of the cell surface receptors, such as EGFR, thus to induce the activation of downstream signaling and invasion (Fig. 7).

Signaling pathway underlying the regulation of vacuolar protein sorting 34 (Vps34) on hepatocellular carcinoma (HCC) invasion. A schematic drawing of the two mechanisms of Vps34 in inhibiting the HCC invasion by regulating the endosome-lysosome system. (A) When Vps34 is sufficient. Vps34 could promote lysosomal juxtanuclear accumulation through Rab7–Rab7-interacting lysosomal protein (RILP) pathway (red arrow), thus less lysosomal peripheral transportation reduced the secretion of exosomes, degradation of extracellular matrix (ECM) and invasion (red cross). (B) When Vps34 is deficient. Vps34 deficiency reduced endocytic degradation (red cross) and promoted the Rab11 mediated endocytic recycling of cell surface receptors, such as epidermal growth factor receptor (EGFR) (red arrow), inducing the activation of downstream signaling and invasion. RTK, receptor tyrosine kinase.
It is well known that Vps34 is an important vesicular sorting protein in endocytosis, autophagy and mTORC1 pathway, which are three pathways relying on the lysosome [3]. Therefore, lysosomes are of importance in mediating Vps34’s biological functions. In our study, we found reduced or even loss of JLA, an interesting phenomenon but meaningful for the cancer cell invasion, in HCC cells. Studies have shown that the peripheral lysosomal membrane proteins are more susceptible to the activated signaling pathway on the cell membrane, such as the PI3K-AKT pathway, which promotes cancer development [19]. Besides, when lysosomes lose JLA and accumulate in the peripheral region of the cells, they are easily fuse with the cytoplasmic membrane and promote exosome secretion, which accelerate the release of hydrolases to the extracellular compartment enabling extracellular matrix degradation and invasiveness [20]. Lysosomal trafficking is regulated by Rab7, an interacting partner of Vps34, and its various downstream effector proteins. Among them, RILP, by associating with Rab7 and recruiting dynein-dynactin motor complexes, leads lysosomes to move along the microtubule towards the minus-end (ie, perinuclear microtubule organizing center), promoting JLA formation [21]. In the present study (as shown in Figs. 3 and 4), when JLA was induced in SMMC-7721 by overexpression of Vps34, invasive ability and exosome markers of HCC cells were significantly reduced, which could be reversed by RILP knocking down.
However, the morphological changes of HepG2 cells with Vps34 downregulation could not be simply explained by lysosomal trafficking changes that found in SMMC-7721 cells with Vps34 overexpression. Since the function of Vps34 is regulated by a variety of regulatory molecules through forming different complexes with Beclin1, for example, complex containing Atg14L medicates the autophagosome formation, and UVRAG complex localizes to early and late endosomes regulating cytokinesis and degradative endocytic traffic [22,23], we speculated that different complexes of Vps34 may contribute to the macrovesicle’s formation in HepG2 with Vps34 knocking down. Additionally, compared to SMMC-7721, HepG2 is a very different cell line with lower invasive abilities and relatively higher Vps34 levels, so whether the characteristic of cell lines is involved in the formation of macrovesicles needs further investigation.
Of note, we found downregulation of Vps34 resulted in the blockage of the endo-lysosomal fusion or degradation in HepG2 cells, rather than the autophagy flux, although the level of Rab7 and LC3 both were lower in carcinoma than in peri-carcinoma of human HCC (Fig. 5, S5 Fig.). Work from the Zhou group also demonstrated that the ablation of Vps34 in the mouse sensory neurons led to impaired endocytosis rather than autophagy [18], and indicated the existence of noncanonical Vps34-independent autophagy pathways which required ATG7 [18]. It may be due to this compensatory mechanism that the dynamic level of LC3, representing autophagy flux, in cells with Vps34 knockdown did not change significantly in our study. Therefore, endocytosis, which share the same substrate disposal machinery (i.e., lysosome) with autophagy, was the dominant impaired pathway in HCC cells when Vps34 was deficient.
In the endocytosis system, accelerated endosomal recycling of cell surface receptors is a well-studied mechanism in cancer invasiveness by enhancing downstream signals, which results in disordered lamellipodia, cell scattering, reduced directional cell migration [24]. Muller et al. [25] demonstrated mutant p53 promoted invasive behavior of cells by enhancing the integrin and EGFR endocytic recycling and constitutive activation of EGFR/integrin signaling, which partly depends on Rab11 and its effector proteins. In our study, we observed Rab11, a marker of recycling endosome that mediates the recovery of the internalized cell surface receptors back to the cell membrane surface [26], was increased in HCC tissue and cells with Vps34 knockdown, accompanied with high EGFR level. Furthermore, Rab11 knockdown reversed the increased invasion, EGFR and p-EGFR level caused by Vps34 knockdown (Figs. 5 and 6). It suggested that enhanced endocytic recycling was one of the mechanisms by which the reduction of Vps34 would increase the invasive abilities. Studies have shown that Rab11 also promoted proliferation and invasion of non-small cell lung cancer cells and bladder carcinoma cells [27,28], which further supported our results.
EGFR phosphorylation residues Tyr 1068 (p-EGFR Y1068) is one of the main phosphorylation sites mediating EGFR dimerization and activation upon EGF stimulation, resulting in cell migration, invasion, and adhesion through ERK/mitogen-activated protein kinase and PI3K/AKT signaling [29,30]. In our study, p-EGFR (Y1068) was increased when Vps34 was downregulated and decreased when Rab11 was koncked down. Besides, AKT and p-AKT (T308), a well-known cancer-promoting signaling downstream of EGFR, were also examined and showed an increase in Vps34 knockdown cells (data not shown). Therefore, enhanced EGFR signaling was a result of enhanced Rab11 mediated recycling and a mechanism by which Vps34 regulated HCC invasion.
In conclusion, we reported the novel finding that Vps34 was significantly decreased in HCC and acted as an invasion suppressor by regulating endosome-lysosome trafficking via Rab7-RILP and Rab11. Therefore, we conclude reasonably that Vps34 could be an invasion suppressor of HCC cells. This study also underscores the importance of the behavioral changes of endosomes and lysosomes, such as amount, size and distribution, which could be used as targets in the future treatment of HCC.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
Notes
Ethical Statement
Ethical approval was obtained from the research ethics committee of Fudan University, and informed consent was obtained from every single patient. The institutional review board of the School of Basic Medical Sciences of Fudan University (IRB no. 20140226-120) approved this study.
Author Contributions
Conceived and designed the analysis: Zhang Z, Wu H.
Collected the data: Zou L, Wang S.
Contributed data or analysis tools: Mao X, Hu Y, Shi J.
Performed the analysis: Qi C, Wu H.
Wrote the paper: Qi C, Wu H.
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Acknowledgments
We thank Dr. Jian-Kang Chen, Associate Professor, Cellular Biology and Anatomy, Augusta University, for his helpful advices and technical support in this study.