Downregulation of HuR Inhibits the Progression of Esophageal Cancer through Interleukin-18
Article information
Abstract
Purpose
The purpose of this study was to investigate the effect of human antigen R (HuR) downregulation and the potential target genes of HuR on the progression of esophageal squamous cell carcinoma (ESCC).
Materials and Methods
In this study, a proteomics assay was used to detect the expression of proteins after HuR downregulation, and a luciferase assay was used to detect the potential presence of a HuR binding site on the 3’-untranslated region (3'-UTR) of interleukin 18 (IL-18). In addition, colony formation assay, MTT, EdU incorporation assay, Western blot, flow cytometry, immunohistochemistry, transwell invasion assay, and wound healing assay were used.
Results
In the present study, we found that the expression of both HuR protein and mRNA levels were higher in tumor tissues than in the adjacent tissues. HuR downregulation significantly suppressed cell proliferation. In addition, the metastasis of esophageal cancer cells was inhibited, while the expression of E-cadherin was increased and the expression of matrix metalloproteinase (MMP) 2, MMP9, and vimentin was decreased after HuR knockdown. Moreover, silencing of HuR disturbed the cell cycle of ESCC cells mainly by inducing G1 arrest. Furthermore, proteomics analysis showed that downregulation of HuR in TE-1 cells resulted in 100 upregulated and 122 downregulated proteins, including IL-18 as a significantly upregulated protein. The expression of IL-18 was inversely regulated by HuR. IL-18 expression was decreased in ESCC tissues, and exogenous IL-18 significantly inhibited the proliferation and metastasis of ESCC cells. The 3'-UTR of IL-18 harbored a HuR binding site, as shown by an in vitro luciferase assay.
Conclusion
HuR plays an important role in the progression of esophageal carcinoma by targeting IL-18, which may be a potential therapeutic target for the treatment of ESCC.
Introduction
Esophageal cancer is one of the most common types of malignancies in the world and has a 5-year survival rate of less than 30% after surgery. Esophageal squamous cell carcinoma (ESCC) is the most common pathological type of esophageal cancer in China. In recent years, although the treatment of esophageal cancer has improved greatly, the overall prognosis of ESCC is still unfavorable [1-3].
Human antigen R (HuR) is a member of the embryonic lethality-abnormal vision (ELAV) gene family and is also known as embryonic lethal abnormal visual-like 1 (ELAVL1) [4]. Being a mRNA binding protein, it is widely involved in the regulation of gene transcription. Of the cis-acting elements of eukaryotic mRNA, the most characteristic is the ARE element (AU-rich element). HuR can increase the stability of its target mRNA by binding to the ARE elements located on the 3'-untranslated region (UTR) of a number of unstable mRNAs. Under normal physiological conditions, HuR is mainly localized to the nucleus. However, under conditions of stress, HuR binds to its target mRNA to form a complex and is shuttled to the cytoplasm. Thus protecting its bound mRNA from degradation [5].
In recent years, there have been reports of abnormally high HuR expression in gastric cancer [6], colorectal cancer [7] and breast cancer tissues [8]. High levels of HuR can induce resistance of pancreatic cancer to gemcitabine. In addition, HuR inhibition increased the sensitivity of pancreatic cancer cells to TRAIL-based therapies and thus supported their re-evaluation as an effective treatment for patients with pancreatic ductal adenocarcinoma [9,10]. Downregulation of HuR can increase the sensitivity of breast cancer line MCF-7 to doxorubicin and increase its rate of apoptosis [11]. Hence, HuR is involved in the biological behavior of multiple tumors and affects their response to chemotherapeutic drugs.
The expression and influence of HuR in the development of esophageal cancer remains unclear. In this study, we investigated the expression of HuR and its functional consequences in ESCC progression. We demonstrated that HuR is significantly overexpressed in ESCC tissues compared to tumor-adjacent tissues. In addition, inhibition of HuR can significantly inhibit the proliferation and metastasis of ESCC cells through interleukin 18 (IL-18).
Materials and Methods
1. Tissue samples
For immunohistochemistry (IHC), 120 ESCC samples and 52 matched tumor-adjacent tissues were collected from 120 patients, as previously reported [12]. Fifteen normal esophageal tissues were obtained from surgical resections of trauma patients. These tissues were obtained postoperatively between 2010-2012 from the Gastrointestinal Center, Jiangyin People’s Hospital, Medical School of University of Southeast of China (Jiangyin, China). All patients gave signed, informed consent for the use of their tissues for scientific research. Ethical approval for the study was obtained from the Jiangyin People’s Hospital. All diagnoses were based on pathological and/or cytological evidence. The histological features of the specimens were evaluated by two senior pathologists according to the World Health Organization classification criteria [12].
2. Cells culture, transfection and treatment
TE-1 and Eca-109 cells were obtained from American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT) supplemented with 10% fetal calf serum and 100 U/mL penicillin–streptomycin at 37°C and a 5% CO2 atmosphere. IL-18 was added to the culture medium at 5 ng/ml according to the manufacturer’s instructions for 48 hours, and then the following experiments were carried out.
3. Generation of HuR downregulated stable cell lines
The human HuR coding region was amplified by polymerase chain reaction (PCR) using a primer pair specific to HuR. The shRNA targeting HuR was obtained from Hanbio Biotechnology (containing RFP, Shanghai, China). The fragments were inserted into the lentivirus (LV) expression vectors LV-HuR shRNA-1 (top strand, GCAGCATTGGTGAAGTTGAATCTGCATTCAAGAGATGCAGATTCAACTTCACCAATGCTGTTTTTT; bottom strand, AAAAAACAGCATTGGTGAAGTTGAATCTGCATCTCTTGAATGCAGATTCAACTTCACCAATGCTGC) and LV-HuR shRNA-2 (top strand, GTACCAGTTTCAATGGTCATAATTCAAGAGATTATGACCATTGAAACTGGT ATTTTTT; bottom strand, AAAAAATACCAGTTTCAATGGCATAATCTCTTGAATTATGACCATTGAAACTGGTAC) and then packaged into viral particles. The particles were used to infect TE-1 cells and Eca-109 cells. The cells were subsequently harvested at 72-hour postinfection and selected in medium containing 3 μg/mL puromycin (Sigma-Aldrich, St. Louis, MO) until all the uninfected cells were killed by puromycin. Stable cell lines were validated using western blot.
4. Immunohistochemistry
Three-micrometer-thick paraffin-embedded tissue sections were deparaffinized and heat-treated with citrate buffer, pH 6.0, for 7 minutes for epitope retrieval. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 15 minutes at room temperature, and nonspecific binding sites were blocked with 4% skim milk for 30 minutes. The sections were then incubated with HuR or IL-18 antibody for 1 hour (1:200, mixed with 2% skim milk to reduce unspecific staining). Biotinylated secondary antibody was then added for 30 minutes. Avidin–biotin–peroxidase complex (Dako LSAB2 system, DAKO Co., Carpinteria, CA) was added, and the color was developed using 3-3′-diaminobenzidine. Counterstaining was performed with hematoxylin. All steps were performed at room temperature.
IHC scoring was based on both cytoplasmic and nucleic staining. The stained sections were scored using the following criteria as reported previously [12]: negative (–), < 10% of the whole tissue mass stained; weakly positive (+1), 10%-25% of the whole tissue mass stained; moderately positive (+2), 25%-75% of the whole tissue mass stained; and strongly positive (+3), > 75% of the tissue showed positive staining [12].
5. Reverse transcriptase-PCR
Total RNA from esophageal tissues was extracted with TRIzol (Invitrogen, Carlsbad, CA) and reverse transcribed to cDNA using an oligo(dT) 12 primer and Superscript II (Invitrogen). The levels of the mRNA from target genes and the internal standard glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were measured by reverse transcriptase-PCR (RT-PCR) in triplicate using a Prism 7500 PCR machine (Applied Biosystems, Foster City, CA). The specific primers were as follows: HuR: forward 5’-ATGAAGACCACATGGCCGAAGACT-3’, reverse 5’-AGTTCACAAAGCCATAGCCCAAGC-3’; GAPDH: forward 5’-AGCCTCAAGATCATCAGCAATGCC-3', reverse 5’-TGTGGTCATGAGTCCTTCCACGAT-3'.
6. Cell proliferation assay
After treatment, the TE-1 or Eca-109 cells were seeded in 96-well plates at a density of 2×104 cells per well. The adherent cells were further cultured in the presence or absence of IL-18. Cell proliferation was measured using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay 48 hours after IL-18 was added. Briefly, 20 μL of the MTT solution (5 mg/mL) and 100 μL complete medium were added to each well and the cells were incubated for 4 hours at 37°C. The medium was then aspirated and 150 μL of dimethyl sulfoxide was added to dissolve the crystals. The optical density was measured at 490 nm using a microplate reader (Bio-Rad, Hercules, CA). The viability index was calculated as the experimental OD value/the control OD value. Three independent experiments were performed in quadruplicates.
7. Colony formation assay
After treatment, the cells were seeded on 6-well plates. After 10 days of incubation at 37°C, the colonies were stained with Giemsa and those with a minimum of 50 viable cells were counted. This process was repeated three times. The cloning efficiency was calculated as a ratio of the number of colonies formed divided by the total number of cells plated times the plating efficiency.
8. EdU incorporation assay
5-Ethynyl-20-deoxyuridine (EdU) is a nucleoside analog of thymidine that is only incorporated into DNA during active DNA synthesis by proliferating cells. After incorporation, a fluorescent molecule that reacts specifically with EdU was added, which made the proliferating cells fluorescent. The Cell-Light EdU DNA cell proliferation kit (RiboBio, Guangzhou, China) was used to determine the proliferation rate of the TE-1 or Eca-109 cells after treatment according to the manufacturer’s instructions. Briefly, the cells were incubated with 50 μM EdU for 2 hours before fixation, permeabilization, and EdU staining. The cell nuclei were stained with Hoechst 33342 at a concentration of 5 μg/mL for 30 minutes, and the cells were examined using a fluorescence microscope (Olympus, Tokyo, Japan).
9. Transwell invasion assay and wound healing migration
We assayed the invasion ability of the cells using 6.5 mm diameter Transwell chambers with 8 μm membranes (Corning Costar, Corning, NY). TE-1 or Eca-109 cells after treatment were added to the upper chambers. The bottom wells were coated with 1 mg/mL Matrigel for the invasion assays and medium containing 20% fetal bovine serum was added to the lower chambers. After 30-hour incubation at 37°C in humidified 5% CO2 atmosphere, the cells in the upper chambers were carefully scraped off using a cotton swab, and those that had migrated to the basal side of the membrane were fixed in 4% paraformaldehyde, stained with Giemsa's stain, and the membranes were mounted and dried at 37°C for 30 minutes. The number of cells invading the Matrigel was counted in three randomly selected fields using an inverted microscope (10× magnification). Each test was performed in triplicate.
For the wound healing migration assay, the cells were seeded onto 6-well plates after treatment and allowed to form a confluent monolayer for 24 hours. The monolayer was scratched with the tip of a 200 μL pipette and the wells were washed twice with phosphate buffered saline (PBS) to remove the floating and detached cells. Then, fresh serum-free medium was added, and photos were taken at 0 and 24 hours to assess cell migration using a microscope (Olympus).
10. Flow cytometric analysis of cell cycle
After treatment, cells in logarithmic growth phase were collected and fixed with 70% precooled ethanol overnight. After staining with propidium iodide (10 μg/mL, Sigma-Aldrich) in the dark for 30 minutes, flow cytometry was performed on the FACSCalibur Flow Cytometer system and the cell cycle distribution was analyzed using the ModFit LT software.
11. Proteomics analysis
According to the manufacturer's instructions, 5×106 TE-1 cells infected with shRNA-HuR-1 or shRNA-NC containing virus were collected and washed three times with PBS. The proteomics analysis was performed as the following steps:
1) Protein extraction
Each samples was sonicated three times on ice using a high intensity ultrasonic processor (Scientz) in lysis buffer (8 M urea, 2 mM ethylenediaminetetraacetic acid, 10 mM dithiothreitol, and 1% protease inhibitor cocktail). The remaining debris was removed by centrifugation at 20,000 g at 4°C for 10 minutes. The protein was precipitated with cold 15% trichloroacetic acid for 4 hours at –20°C. After centrifugation at 4°C for 3 minutes, the supernatant was discarded. The remaining precipitate was washed three times with cold acetone. The protein was redissolved in buffer (8 M urea, 100 mM triethyl ammonium bicarbonate [TEAB], pH 8.0), and the protein concentration in the supernatant was determined with 2-D Quant kit according to the manufacturer’s instructions.
2) Trypsin digestion
For digestion, the protein solution was reduced with 10 mM DTT for 1 hour at 37°C and alkylated with 20 mM iodoacetamide for 45 minutes at room temperature in darkness. For trypsin digestion, the protein sample was diluted by adding 200 mM TEAB to reduce the urea concentration to less than 2 M. Finally, trypsin was added at 1:50 trypsinto-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for a second digestion for 4 hours. Approximately 100 μg protein for each sample was digested with trypsin for the following experiments.
3) Tandem mass tags labeling
After trypsin digestion, the peptides were desalted by Strata X C18 SPE column (Phenomenex) and vacuum-dried. The peptides was reconstituted in 1 M TEAB and processed according to the manufacturer’s protocol for 6-plex tandem mass tags (TMT) kit. Briefly, one unit of TMT reagent (defined as the amount of reagent required to label 100 μg of protein) was thawed and reconstituted in 24 μL acetonitrile (ACN). The peptide mixtures were then incubated for 2 hours at room temperature and pooled, desalted and vacuum-dried.
4) High-performance liquid chromatography fractionation
The samples were fractionated by high pH reverse-phase high-performance liquid chromatography using Agilent 300Extend columns (5 μm particles, 4.6 mm ID, 250 mm length; Agilent Technologies, San Jose, CA). Briefly, the peptides were first separated with a gradient of 2% to 60% ACN in 10 mM ammonium bicarbonate pH 10 over 80 minutes into 80 fractions. Then, the peptides were combined into 18 fractions and dried by vacuum centrifuging.
5) Liquid chromatography-tandem mass spectrometry analysis
The peptides were dissolved in 0.1% FA, directly loaded onto a reversed-phase precolumn (Acclaim PepMap 100, Thermo Fisher Scientific, Waltham, MA). Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC, Thermo Fisher Scientific). The elution gradient comprised of an increase from 4% to 22% solvent B (0.1% FA in 98% ACN) over 26 minutes, 22% to 35% in 8 minutes before climbing to 80% in 3 minutes, and finally holding at 80% for the last 3 minutes, all at a constant flow rate of 350 nL/min on an EASY-nLC 1000 UPLC system. The resulting peptides were analyzed by Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific).
The peptides were subjected to a nanospray ionization source followed by tandem mass spectrometry (MS/MS) in Q Exactive coupled online to the UPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using a normalized collision energy setting of 28 and the ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above the threshold ion count of 1×104 in the MS survey scan with 30.0-second dynamic exclusion. The applied electrospray voltage was 2.0 kV. Automatic gain control was used to prevent overfilling of the Orbitrap. Fifty thousand ions were accumulated for the generation of the MS/MS spectra. For MS scans, the m/z scan range was 350 to 1800, and the fixed first mass was set as 100 m/z.
6) Database search
The resulting MS/MS data were processed using the Mascot search engine (v.2.3.0). Tandem mass spectra were searched against the Swiss-Prot Homo sapiens database. Trypsin/P was specified as the cleavage enzyme allowing up to two missing cleavages. The mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethyl on Cys were specified as fixed modifications and oxidation on Met were specified as variable modifications. For protein quantification, TMT 6-plex was selected in Mascot. False discovery rate was adjusted to < 1% and peptide ion score was set ≥ 20. In addition, the bioinformatics analysis was performed according to the operation process of the manufacturer (PTM-Biolabs, Hangzhou, China).
12. Luciferase assay
A luciferase reporter containing the full-length 3’-UTR of IL-18 (pGL3-UTR-WT) and a mutant reporter plasmid with a mutation of the HuR binding site (pGL3-UTR-1Del1) were constructed by Synbio Tech Ltd. (Suzhou, China). For luciferase assay, 500 ng of the reporter vector was cotransfected into cells with 500 ng synthetic RNA in a 24-well plate. Thirty nanograms of pRL-TK (Promega, Madison, WI) was also transfected to correct for transfection efficiency. The luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega). The ratio of firefly luciferase to Renilla luciferase activities was used to determine promoter activities.
13. Western blotting
The proteins in cell lysates or exosomes were resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane, which was then blocked with PBS/Tween-20 containing 5% skim milk. The membranes were incubated with antibodies against HuR, IL-18 (Abcam, Cambridge, MA), matrix metalloproteinase 2 (MMP2), matrix metalloproteinase 9 (MMP9), E-cadherin, vimentin (Cell Signaling Technology, Beverly, MA) and β-actin. Goat anti-mouse and goat anti-rabbit secondary antibodies were purchased from Beyotime (Nantong, China).
14. Statistical analyses
Data are expressed as the mean±standard error of mean of at least three independent experiments. The data were analyzed by one-way analysis of variance (ANOVA). Statistical analyses were performed using PRISM ver. 6.0 (GraphPad Software, San Diego, CA). Statistical significance was considered for p-value < 0.05. The relative mRNA and protein expression levels were analyzed using the ImageJ image analysis software (NIH, Bethesda, MD).
Results
1. Both protein and mRNA levels of HuR were significantly higher in the tumors than in the adjacent tissues
To test the hypothesis that tumor-adjacent tissues express different subsets of the HuR gene, we measured HuR expression by immunohistochemistry. Immunohistochemical staining was performed on 120 ESCC tissues and 52 matched tumor-adjacent tissues. The histopathological features of the patients were described in a previous publication from our research group [12]. HuR was located in both the nucleus (green arrow) and cytoplasm (red arrow) of cells of the ESCC tissues, whereas very weak staining of HuR was observed in the cytoplasm of the tumor-adjacent tissues (red arrow in Fig. 1A). Additionally, four specimens were selected for RT-PCR, and the results show that the expression of HuR mRNA in esophageal cancer tissues was significantly higher than the matched tumor-adjacent normal tissues (Fig. 1B).

Both human antigen R (HuR) protein and mRNA levels were significantly higher in tumor tissues than that in the adjacent tissues. (A) Representative images of the different staining patterns are presented. (B) RNA was extracted from four tumor tissues and matched normal tissues, and HuR mRNA expression was detected by polymerase chain reaction. ESCC, esophageal squamous cell carcinoma; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
2. Downregulation of HuR inhibited proliferation and disturbed the cell cycle of esophageal cancer cells
Western blot analysis of HuR expression in esophageal cancer cells showed that compared to the cells infected with shRNA-NC, HuR expression in both TE-1 and Eca-109 cells infected with shRNA-1 and shRNA-2 was reduced by approximately 3 to 5 times (Fig. 2A). Cell growth monitored by MTT assay showed that HuR downregulation can inhibit the proliferation of TE-1 and Eca-109 by approximately 10%-15% (Fig. 2B). EdU staining was performed to investigate the effect of HuR on the proliferation of esophageal cancer cells. Downregulation of HuR decreased the proliferation rate compared to the control by approximately 10%-15% (Fig. 2C). Colony formation assay showed that the clonogenicity of esophageal carcinoma was significantly reduced after HuR silencing by approximately 10%-20% (Fig. 2D). Furthermore, the cell cycle of esophageal cancer was measured by flow cytometry. The results showed that the G1 phase was significantly prolonged and the S phase was significantly shortened by downregulation of HuR (Fig. 3). Taken together, these results showed that HuR downregulation could inhibit the proliferation and colony forming efficiency of esophageal cancer cells by approximately 15%. In addition, the esophageal cancer cells were arrested in the G1 phase, leading to a decrease in proliferation.
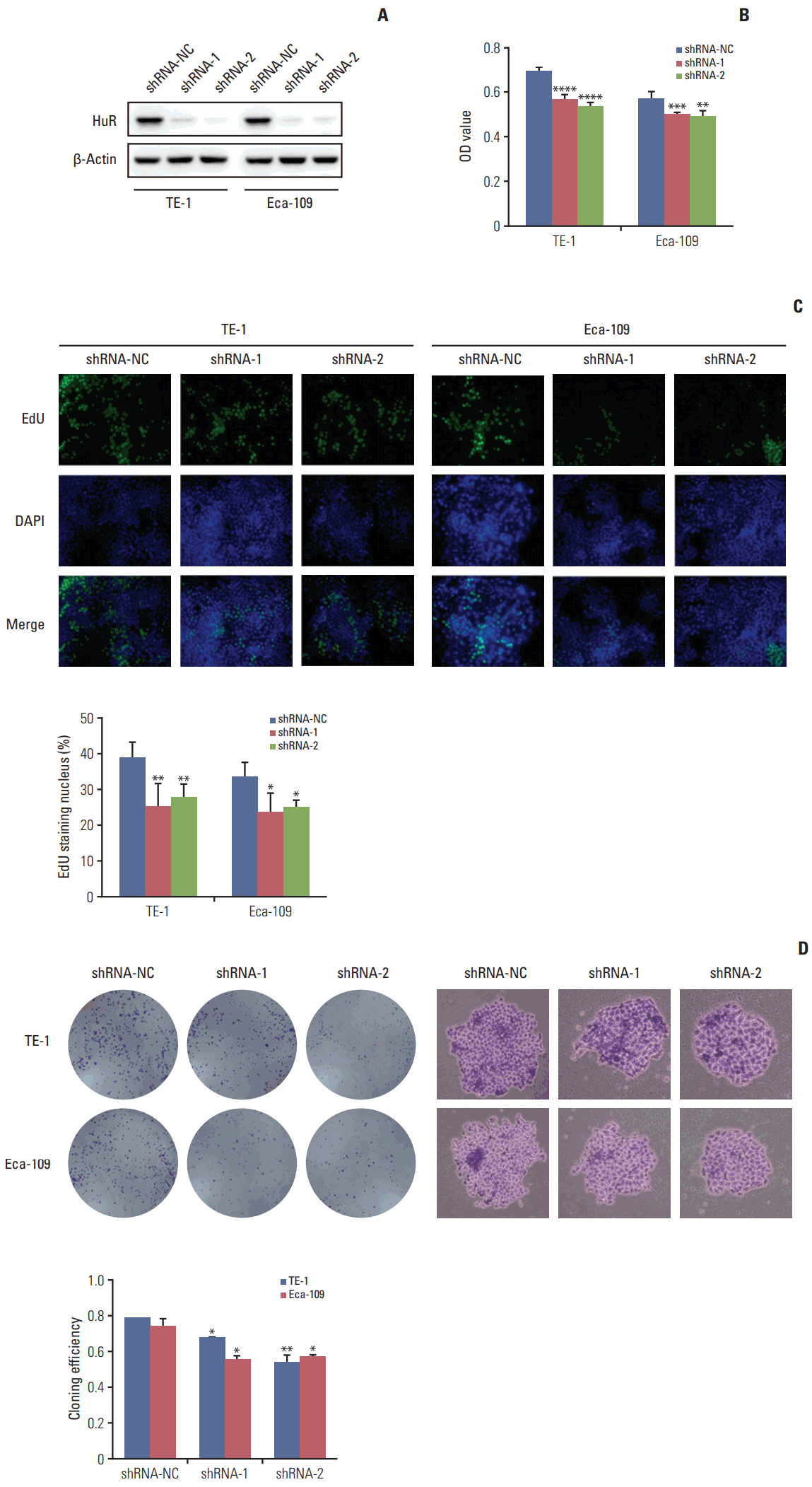
Downregulation of human antigen R (HuR) inhibited cell proliferation. (A) Western blot was used to detect the expression of HuR in esophageal cancer cells after shRNA transfection. (B) The effect of HuR on the viability of the TE-1 and Eca-109 cells infected with the HuR knockdown virus. The cells were seeded in a 96-well plate and incubated for 48 hours before being analyzed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. (C) The proliferating TE-1 and Eca-109 cells were labeled with 5-ethynyl-20-deoxyuridine (EdU). The Click-iT reaction revealed EdU staining (green). The nuclei were stained with Hoechst 33342 (blue). The images are representative of the results obtained. (D) Colony formation assays of TE-1 and Eca-109 cells after treatment. After 10 days, the cells were stained with crystal violet. Colonies consisting of more than 50 cells were counted. The data are presented as the mean±standard error of mean (n=3). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, compared to the negative control.
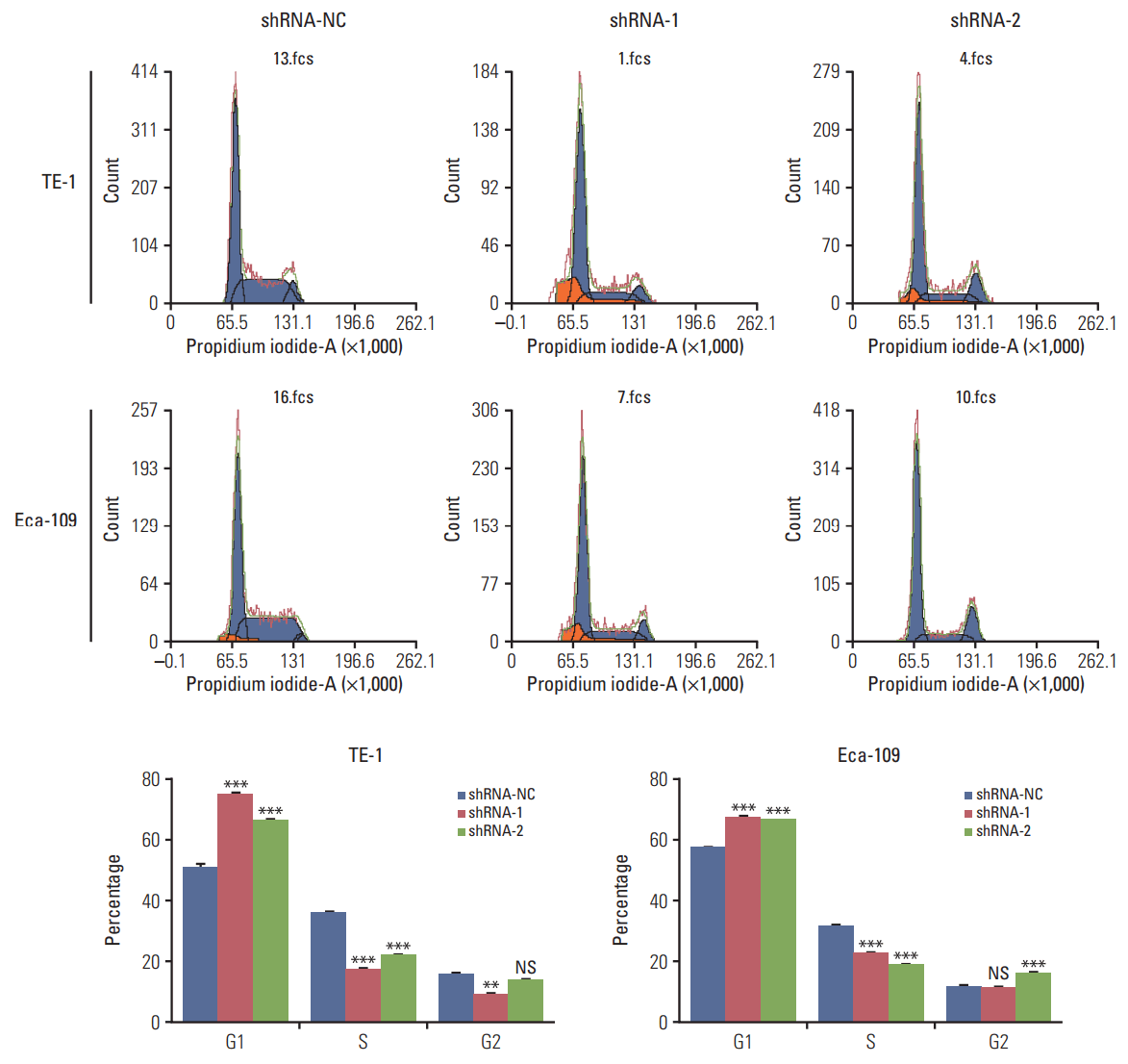
Downregulation of human antigen R (HuR) disturbed the cell cycle of esophageal squamous cell carcinoma cells. TE-1 and Eca-109 cells infected with the HuR knockdown virus were stained with propidium iodide in preparation for flow cytometry with the FACSCalibur system as described in Materials and Methods. (**p < 0.01 and ***p < 0.001).
3. Downregulation of HuR inhibited invasion and metastasis of ESCC cells
The invasion of esophageal cancer cells was measured by Transwell chambers. The results showed that downregulation of HuR can significantly reduce TE-1 and Eca-109 invasion by approximately three times compared to the control group (Fig. 4A). Wound healing assays were used to detect the migration of the two cell types, and the results were consistent with the findings from the invasion assay (Fig. 4B). Western blot analysis showed that downregulation of HuR decreased the expression of the metastasis effectors MMP2, MMP9, vimentin while increasing the E-Cadherin expression (Fig. 4C). Overall, downregulation of HuR significantly inhibited the invasion and metastasis of esophageal cancer cells.

Downregulation of human antigen R (HuR) inhibited cell invasion and metastasis in esophageal squamous cell carcinoma cells. (A) Migration of the cells was assessed using a transwell-based assay. The TE-1 and Eca-109 cells infected HuR knockdown virus were seeded in the upper chamber and incubated for 30 hours at 37°C. The cells in the upper chamber were carefully scraped off using a cotton swab, and those that had migrated to the basal side of the membrane were fixed in 4% paraformaldehyde and stained with Giemsa. The membranes were mounted and dried at 37°C for 30 minutes. (B) Wound healing was observed 48 hours after the cells treated. The open wound area after incubation was normalized to the initial wound area. The data are presented as the mean±standard error of mean normalized to the control cells (**p < 0.01 and ***p < 0.001). (C) TE-1 and Eca-109 cells infected with the HuR knockdown virus were subjected to Western blot analysis for β-actin, HuR, matrix metalloproteinase (MMP) 2, MMP9, E-cadherin, and vimentin.
4. Identification of HuR regulated proteins using a quantitative proteomics approach
Unlike most studies that perform correlation analysis by DNA and mRNA profiling, here we analyzed the relationship between the DNA and the protein profiles by proteomics experiments. The results showed that compared to the control group downregulation of HuR in TE-1 cells resulted in 100 proteins being upregulated and 122 proteins being downregulated (S1 Fig. A). The results included upregulation of many important proteins such as E3 ubiquitin-protein ligase TRAF7 (tumor necrosis factor receptor-associated factor 7). TRAF7 is a newly discovered member of the TRAF family, which has been shown to be involved in the genesis and progression of several human cancers, putting it in the spotlight as a novel tumor suppressor protein [13]. Among the downregulated proteins, exportin-5 has also been an active research topic recently. Recent studies have shown that Exportin-5 plays an important role in tumorigenesis. A study by Ott et al. [14] found that a high abundance of exportin-5 in the melanoma leads to enhanced survival, proliferation and metastasis and thereby supports the aggressiveness of the cancer [14]. Studies have reported that endogenous IL-18 can affect hepatic metastasis by upregulating melanoma cell adhesion to HSE cells and tumor growth, implicating a possible antimetastatic benefit of neutralizing IL-18. In addition, IL-18 has been shown to promote tumorigenesis, metastasis and angiogenesis [15,16]. Thus, IL-18 can have a significant effect on tumor progression (S2 and S3 Tables). The quantifiable proteins in this study were divided into four categories according to their ratios: Q1 (0 < ratio < 1/1.3, p < 0.05), Q2 (1/1.3 < ratio < 1/1.2, p < 0.05), Q3 (1.2 < ratio < 1.3, p < 0.05), and Q4 (ratio > 1.3, p < 0.05). Then, the proteins from these four categories were plotted for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment-based clustering analysis, and 21 processes or diseases were included (S1 Fig. B).
To obtain relevant pathway from the proteomics analyses, KEGG-pathway database was used. This is a collection of manually drawn pathway maps representing our knowledge of the molecular interactions and reaction networks for genetic information processing. The KEGG pathway-based enrichment analysis revealed 20 upregulated proteins and 11 downregulated proteins in cells infected with shRNA-1 vs. shRNANC (S1 Fig. C and D).
5. Downregulation of HuR promoted IL-18 expression by binding to its 3′-UTR
The proteomics results showed that IL-18 expression was significantly upregulated after HuR downregulation. Therefore, IL-18 was selected as the target of our study (S2 Table). Western blot analysis showed that HuR knockdown could significantly upregulate the expression of IL-18 (Fig. 5A). Moreover, we found that the 3′-UTR of IL-18 mRNA of harbors an AU-rich element, which is likely to be a binding site for HuR. To explore the possibility that HuR regulates IL-18 through its 3′-UTR, two luciferase constructs containing the IL-18 promoter with or without HuR binding sites (pGL3-IL-18-WT and pGL3-IL-18-Del1, respectively) (Fig. 5B) were constructed. Luciferase assays showed that downregulation of HuR enhanced the promoter activities in both TE-1 and Eca-109 cells (Fig. 5C). These results indicated that the 3′-UTR of IL-18 harbors a binding site for HuR and that HuR regulates the expression of IL-18 protein in esophageal cancer cells.

Human antigen R (HuR) negatively regulates the expression of interleukin 18 (IL-18) in esophageal cancer cells. (A) Western blot was used to evaluate the expression of IL-18 in esophageal carcinoma cells in which HuR was downregulated. (B) The diagram indicates the relative positions of the transcription factor HuR on the IL-18 promoter. Truncated promoters were cloned upstream of the firefly luciferase reporter gene. (C) Luciferase reporters containing IL-18 promoters were transfected into TE-1 and Eca-109 cells. After 24-hour incubation, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). NS, not significant. **p < 0.01, ****p < 0.0001.
6. IL-18 inhibited the proliferation and invasion of esophageal cancer cells
IL-18 (5 ng/mL) was added to the cell culture medium. An MTT assay was performed 48 hours later. After 10 days, the cells were fixed and stained with Giemsa, and the number of clones was calculated. A cluster of greater than or equal to 50 cells was defined as a clone. The clone formation rate is equal to the number of clones formed divided by the number of cells inoculated. IL-18 significantly inhibited tumor cell survival (Fig. 6A) and clone formation (Fig. 6B). The cell cycle distribution of esophageal cancer cells treated with IL-18 for 48 hours was evaluated by flow cytometry. Exogenous IL-18 had no effect on the cell cycle of esophageal cancer cells (Fig. 6C). The same number of cells from treated and untreated (3×104) were inoculated in the upper chamber of a Transwell. After 30 hours, the cells were fixed, stained with Giemsa, and photographed under a microscope (100×). The results showed that IL-18 treatment significantly reduced the number of invasive cells (Fig. 6D). Wound healing assays showed that IL-18 significantly inhibited the migration of esophageal cancer cells and the scratched area closed more slowly (Fig. 6E). Western blot analysis showed that exogenous IL-18 inhibited the expression of the metastasis effectors MMP2 and MMP9 but had no effect on HuR (Fig. 6F). Taken together, our results showed that IL-18 significantly inhibited the proliferation and metastasis of esophageal cancer cells.
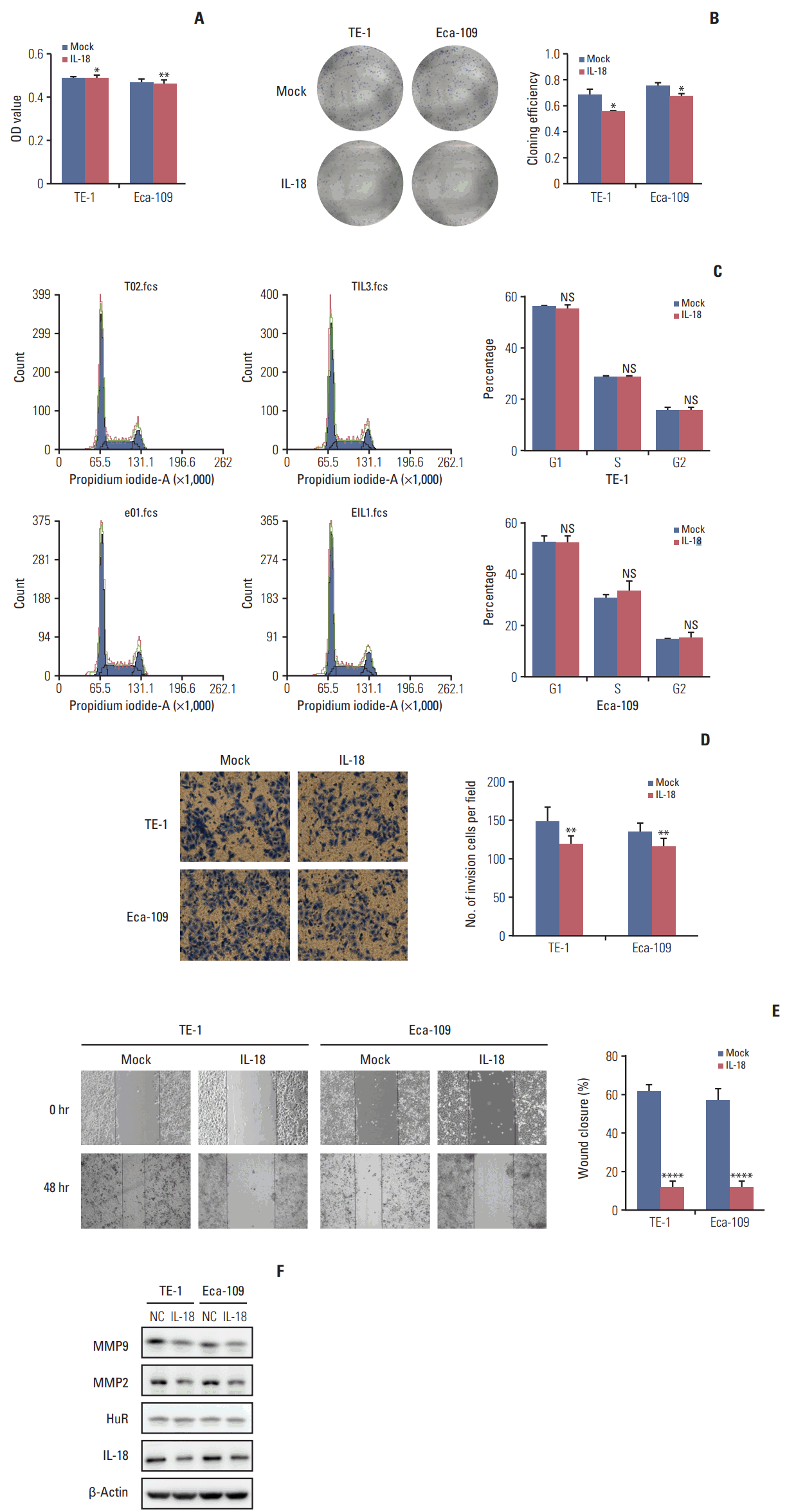
Interleukin 18 (IL-18) inhibited proliferation and invasion of esophageal squamous cell carcinoma cells. (A) The effect of human antigen R (HuR) on the viability of TE-1 and Eca-109 cells seeded in 96-well plates and treated with IL-18 (5 ng/mL) for 48 hours were analyzed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (*p < 0.05, **p < 0.01). (B) Colony formation assays of TE-1 and Eca-109 cells treated with IL-18 (5 ng/mL). After 10 days, the cells were stained with crystal violet. Colonies consisting of more than 50 cells were counted. The data are presented as the mean±standard error of mean (SEM) (n=3). *p < 0.05 compared to the negative control. (C) TE-1 and Eca-109 cells were treated with IL-18 for 48 hours, and stained with propidium iodide in preparation for flow cytometry with the FACSCalibur system as described in Materials and Methods. (D) Migration of cells was assessed using a transwell assay. The TE-1 and Eca-109 cells treated for 48 hours with IL-18 were seeded in the upper chambers and incubated for 30 hours at 37°C. Then, the cells in the upper chambers were carefully scraped off using a cotton swab, and the ones that had migrated to the basal side of the membrane were fixed in 4% paraformaldehyde and stained with Giemsa, and the membranes were mounted and dried at 37°C for 30 minutes (**p < 0.01). (E) Wound healing was observed 48 hours after the cells were treated with IL-18, and the open wound area after incubation was normalized to the initial wound area. The data are presented as the mean±SEM normalized to the control cells (****p < 0.0001). (F) Protein extracts from TE-1 and Eca-109 cells treated for 48 hours with IL-18 were subjected to Western blot analysis for HuR, IL-18, matrix metalloproteinase (MMP) 2, and MMP9.
7. Low expression of IL-18 in esophageal cancer tissues
To determine the role of IL-18 in ESCC development, we investigated the expression of IL-18 in 120 clinical ESCC tissues and 52 matched tumor-adjacent tissues. The expression of endogenous IL-18 between human ESCC tissues and paired adjacent non-tumor tissues was analyzed by IHC, and the stained sections were scored. As shown in Fig. 7A, IL-18 was located in both the nucleus (green arrow in Fig. 7A) and cytoplasm (red arrow in Fig. 7A) of cells of the tumor-adjacent tissues, whereas very weak staining of IL-18 was observed in the cytoplasm of the ESCC cells (red arrow in Fig. 7A). IL-18 expression in tumor-adjacent tissues was significantly different from that of the ESCC tissues (Fig. 7B).

Low expression of interleukin 18 (IL-18) in esophageal cancer tissues. (A) Representative images of the different staining patterns are presented. (B) Box plots depicting IL-18 staining scores as assessed by immunohistochemisty (IHC) in tumors and adjacent tissues in our series of 52 paired esophageal squamous cell carcinoma (ESCC) and tumor adjacent tissue samples. ****p < 0.0001.
Discussion
HuR has been identified as one of a small number of RNA-binding proteins. HuR regulates the process of mRNA export from the nucleus to the cytosol after transcription, stabilizes the mRNA and promotes translation. HuR-linked targets include oncogenes [17], tumor suppressor genes [18] and modulators of tumorigenesis [19]. HuR is involved in different biological functions of tumor cells, including transcription, cell cycle, inflammatory response, cell adhesion and apoptosis, by regulating the expression of different genes [20], which is consistent with our research results. Thus, HuR may serve as a target for cancer therapy. In a study by Muralidharan et al. [21], it was shown that HuR-specific siRNA contained in a folate-targeted lipid nanoparticle (HuR-FNP) was capable of delivering HuR targeting siRNA to folate receptor alpha–expressing tumor cells to produce cell-specific knockdown of HuR and HuR-regulated target proteins. However, this resulted in minimal cytotoxicity to normal cells. These in vitro study results provide an impetus for testing HuR-FNP in vivo either as a monotherapy or in combination therapy with conventional chemotherapeutics and other molecular targeted therapies for lung cancer. A study by Leijon et al. [22] showed that increased expression of HuR protein is associated with metastatic potential of paragangliomas and pheochromocytomas, and cyclooxygenase 2 (COX-2) was identified as a target of HuR. In many malignancies, HuR expression is associated with high grade, poor prognosis, and large tumor size [23]. However, a few opposing results have also reported. High levels of HuR expression have also been shown to be connected with more favorable prognosis in breast carcinoma [24]. In this study, we found that the expression of HuR in esophageal carcinoma was significantly higher than that in adjacent tissues. HuR downregulation could significantly inhibit the proliferation, invasion, migration, and cell cycle of esophageal cancer cells.
As previously mentioned, HuR can regulate the transcription and protein expression of many genes. Proteomics analysis was the most important method used in this study. In our results, we found a number of important proteins whose expression or function was closely related to HuR. Zhang et al. [25] found that HuR was required for heregulininduced nuclear translocation of the nuclear factor κB (NF-κB) subunit p65, which contains both classical and noncanonical nuclear localization signals. HuR thus regulates the transcriptional activities of both nuclear retinoic acid receptor and NF-κB. Therefore, HuR plays a central role in regulating nuclear import of proteins [25]. In our proteomics analysis, we found that downregulation of HuR could enhance the expression of NF-κB P100 subunit, which results in the reduction of NF-κB nuclear translocation. Chu et al. [26] identified a novel post-transcriptional mechanism by which HuR regulated insulin-like growth factor-I receptor (IGF-IR) expression through mRNA stabilization. They showed that HuR acted as a translation repressor of IGF-IR mRNA by binding at the 5’-UTR and that ectopic HuR expression increased IGF-IR mRNA stability and protein expression. This was consistent with our proteomics result that IGF-1 protein was significantly increased in esophageal carcinoma after downregulation of HuR.
IL-18 has been shown to play an important role in tumorigenesis and cancer progression. Li et al. [27] found that breast cancer cell migration and invasion are promoted by leptin via IL-18 expression and secretion, which is regulated by the phosphoinositide 3-kinase–AKT/ATF-2 signaling pathway. It is reported that high levels of the inflammatory cytokine IL-18 in the circulation of patients with hepatocellular carcinoma correlated with poor prognosis. IL-18 exerted inflammation-dependent tumor-suppressive effects largely by promoting differentiation, activity and survival of tumor-infiltrating T cells [28]. In glioma cells, the involvement of HuR in the regulation of IL-18 has also been reported, which is consistent with our finding in ESCC cells. Ko et al. [29] found that IL-18 could be a therapeutic target for treating acute myeloid leukemia. They showed that IL-18 induced the shuttling of HuR and insulin-like growth factor mRNA binding protein 3 from the nucleus to the cytoplasm and facilitated their interaction. In turn, this complex bound to the 3’-UTR of the COX-2 mRNA and enhanced its stability [29]. In this study, we showed that the downregulation of HuR promoted IL-18 protein expression in esophageal TE-1 and Eca-109 cells. Exogenous IL-18 significantly inhibited cell proliferation and invasion. In addition, we showed that the 3′-UTR of IL-18 harbors a HuR binding site. The expression of IL-18 was negatively correlated with the expression of HuR in cancer tissues. In summary, we demonstrate that IL-18 may be a novel and direct target of HuR in esophageal cancer.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (http://www.e-crt.org).
Notes
Conflict of interest relevant to this article was not reported.