Efficacy and Safety of Regorafenib in Korean Patients with Advanced Gastrointestinal Stromal Tumor after Failure of Imatinib and Sunitinib: A Multicenter Study Based on the Management Access Program
Article information
Abstract
Purpose
The aim of this study was to confirm the efficacy and safety of regorafenib for advanced gastrointestinal stromal tumors (GISTs) reported in the GRID phase III trial in Korean patients.
Materials and Methods
Fifty-seven Korean patientswith advanced GISTwho experienced both imatinib and sunitinib failure were enrolled in the management access program between December 2012 and November 2013 and treated with regorafenib (160 mg orally once daily in a 3 weeks on/1 week off).
Results
None of the patients achieved a complete or partial response while 25 patients (44%) showed stable disease for ≥ 12 weeks. With a median follow-up of 12.7 months (range, 0.2 to 27.6 months), the median progression-free survival and overall survival were 4.5 months (95% confidence interval [CI], 3.8 to 5.3) and 12.9 months (95% CI, 8.1 to 17.7), respectively. Interestingly, 15 patients (26%) experienced an exacerbation of their cancer-related symptoms (abdominal pain in eight and abdominal distension in five) during the rest period for regorafenib, but all were ameliorated upon the resumption of regorafenib. The most common grade 3 or 4 adverse event was a hand-foot skin reaction (25%). The regorafenib dose was reduced in 44 patients (77%) due to toxicity, which manifested mainly as a hand-foot skin reaction (n=31).
Conclusion
This study confirmed the efficacy and safety of regorafenib for advanced GIST after imatinib and sunitinib failure in Korean patients. Considering the exacerbation of the cancer-related symptoms observed during the rest periods, further exploration of the continuous dosing schedule of regorafenib is warranted in future clinical trials.
Introduction
Gastrointestinal stromal tumors (GIST) are the most common sarcomas of the gastrointestinal tract [1]. Activating mutations of KIT are observed in approximately 85% of GISTs and activation of the KIT signaling pathway is enhanced, which is responsible for the emergence of malignant GISTs [2]. Mutations in the genes encoding platelet-derived growth factor receptor α (PDGFRA) were later suggested to be another mechanism underlying the malignant GIST tumorigenesis observed [3]. These genetic mechanisms in GISTs are responsible for and can predict their response to imatinib, which was the first tyrosine kinase inhibitor (TKI) approved for the treatment of GIST that has activity against KIT, PDGFRA, and ABL [4]. Although imatinib has been shown to improve the progression-free survival (PFS) and overall survival (OS) remarkably, and has shown partial response (PR) and stable disease (SD) in 54% and 28% of patients with advanced or metastatic GISTs, respectively [5,6], most patients eventually develop resistance to imatinib and progress [7]. Sunitinib, a multitarget TKI that has shown a longer PFS (27 weeks) compared to the placebo arm (6 weeks) in a randomized phase III trial [8], was approved for the second-line treatment in patients with GIST experiencing imatinib failure; however, resistance to sunitinib also developed in most patients [9]. Therefore, a new TKI is needed to overcome the drug resistance as treatment for patients with disease progression.
Regorafenib is a multikinase inhibitor that has demonstrated activity against a range of targets, including KIT, PDGFRA, vascular endothelial growth factor receptor, RAF1, BRAF, RET, and fibroblast growth factor receptor in in vitro analyses [10]. In a multicenter phase II trial of regorafenib in 34 patients with GIST, who had suffered treatment failure after using imatinib and sunitinib for a median of 21 months (range, 1 to 79 months) and 13 months (range, 2 to 55 months), respectively, four patients showed a PR and 22 patients showed SD [11]. In an international, multicenter, randomized, placebo-controlled, phase III trial (GRID), 199 patients with metastatic or unresectable GISTs were randomized to either regorafenib or a placebo after the failure of imatinib and sunitinib [12]. The median PFS was 4.8 months for regorafenib, which was significantly longer than that of 0.9 months in the placebo group. The most common side effects of regorafenib were hand-foot skin reactions (HFSRs), diarrhea, hypertension, and fatigue. Based on that study, regorafenib was approved by the Food and Drug Administration in February 2013 for metastatic or unresectable GIST after imatinib and sunitinib. Subgroup analysis evaluating Japanese patients in the phase III GRID trial reported that HFSR and maculopapular rash were observed more frequently in Japanese patients than in the overall population [13].
The present study examined the efficacy and safety of regorafenib in Korean patients with advanced GIST who either progressed or could not tolerate the toxicity of imatinib and sunitinib.
Materials and Methods
1. Study design and participants
A total of 57 patients with advanced GIST that was refractory or intolerant to imatinib and sunitinib treatments from three Korean institutions (Asan Medical Center, Samsung Medical Center, and Seoul National University Hospital) were enrolled in the management access program (MAP) for regorafenib between December 2012 and November 2013. The purpose of the MAP was to provide unapproved medications for patients in need before commercial access. This is a prospective study, where the patients enrolled in MAP were followed up with planned visits or an additional phone call when necessary and the data for toxicity, response, medication compliance, and survival were collected prospectively.
The eligibility criteria for the MAP included age ≥ 18 years, a histologically confirmed metastatic and/or unresectable GIST, prior failure (progression or intolerance) of at least imatinib and sunitinib, an Eastern Cooperative Oncology Group (ECOG) performance status of ≤ 1, and an adequate bone marrow, liver, and renal function as assessed in a laboratory test. In addition, before entering the program, women of childbearing age and men had to agree to use adequate contraception until at least 8 weeks after the last dose of regorafenib. The key exclusion criteria included prior treatment with regorafenib, congestive heart failure with a ≥ New York Heart Association class 2, unstable angina (angina symptoms at rest) or new-onset angina (within the last 3 months), cardiac arrhythmias requiring anti-arrhythmic therapy (beta blockers or digoxin were permitted), uncontrolled hypertension defined as a systolic blood pressure > 140 mm Hg or diastolic pressure > 90 mm Hg despite the optimal medical management, and a major surgical procedure, open biopsy, or significant traumatic injury within 28 days prior to starting regorafenib.
2. Medications
The patients received oral regorafenib at 160 mg once daily in a 3 week on/1 week off schedule, every 4 weeks. The medication was continued until unmanageable toxicity, disease progression, or consent withdrawal had occurred. Regorafenib was allowed to continue after disease progression if a clinical benefit was evident to the treating physician. Medication was delayed or the dose was reduced to manage any clinically significant drug-related toxicity according to the investigator’s decision. Dose re-escalation was allowed after resolution of toxic effects.
3. Response and toxicity evaluation
The tumor response was assessed using computed tomography or MRI after the first cycle, and then after every two cycles of study medication (i.e., 4 weeks, 12 weeks, and 20 weeks after commencing regorafenib). The investigators evaluated the tumor responses according to the Response Evaluation Criteria in Solid Tumors ver. 1.1.
History taking, physical examination, checking vital signs, and laboratory evaluation were performed every cycle to monitor the toxicity and safety. The toxicity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events ver. 4.0.
4. Statistical analysis
The PFS (time from study entry to disease progression or death from all-causes before progression) and OS (time from study entry to death from all-causes) were calculated using the Kaplan-Meier method and compared among the different subgroups using a log-rank test. Multivariate analysis using the Cox proportional hazard model was performed to obtain the hazard ratio (HR) and 95% confidence interval (CI) values. Variables that showed statistical significance in univariate analysis and/or were considered to be significant in previous studies were included in multivariate analysis. A p-value < 0.05 was considered significant. Statistical analysis was performed using the SPSS ver. 21.0 for Windows (IBM Co., Armonk, NY).
Results
1. Baseline characteristics
Table 1 lists the baseline clinical characteristics of the 57 enrolled patients. Five patients with an ECOG performance status of 1 at enrollment experienced a worsening of their performance status, which was probably caused by rapid tumor progression, and a showed an ECOG performance status of 2 when starting regorafenib. The median time using imatinib and sunitinib was 40 months (range, 9 to 114 months) and 8 months (range, 1 to 44 months), respectively. At the data cutoff date (July 15, 2015), 14 patients (25%) were continued on regorafenib. Of these, eight were receiving regorafenib after disease progression due to an evident clinical benefit. The median time using regorafenib was 4.7 months (range, 0.9 to 27.1 months). Forty-three patients (75%) discontinued regorafenib due to disease progression (n=27), death (n=10), adverse events (n=4), or consent withdrawal (n=2).
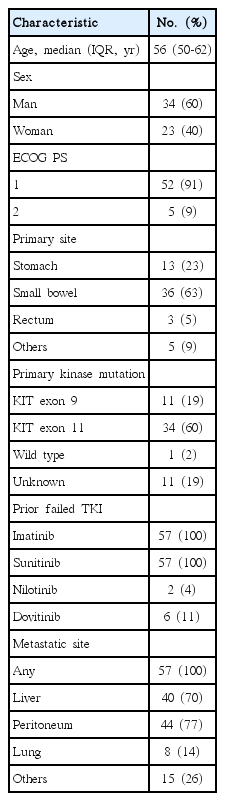
Baseline patient characteristics
2. Efficacy
The first tumor response assessed after commencing regorafenib was SD, progressive disease, and not evaluable in 46, six, and five patients, respectively. Twenty-five patients (44%) had SD for ≥ 12 weeks, and none achieved a complete response or PR. Ten patients could not be evaluated for the response as they had died before 12 weeks of tumor assessment (n=7), resection of the target lesions due to bleeding (n=1) and perforation (n=1), and radiofrequency ablation for liver metastasis (n=1).
Fig. 1 presents the Kaplan-Meier curves of the PFS and OS outcomes. With a median follow-up of 12.7 months (range, 0.2 to 27.6 months), the median PFS was 4.5 months (95% CI, 3.8 to 5.3). The median OS was 12.9 months (95% CI, 8.1 to 17.7).

Progression-free survival (PFS) (A) and overall survival (OS) (B) for the entire study population.
Patients with a liver metastasis had a significantly shorter PFS and OS according to both univariate (Fig. 2) and multivariate (Table 2) analysis. No differences in the PFS and OS were evident between patients with a primary KIT exon 11 mutation, a primary KIT exon 9 mutation or wild type in univariate analysis (Fig. 2) or multivariate Cox-proportional hazard analysis (PFS: HR, 0.62; 95% CI, 0.26 to 1.50; OS: HR, 0.83; 95% CI, 0.32 to 2.13). All 36 deaths encountered during the present study were due to disease progression.
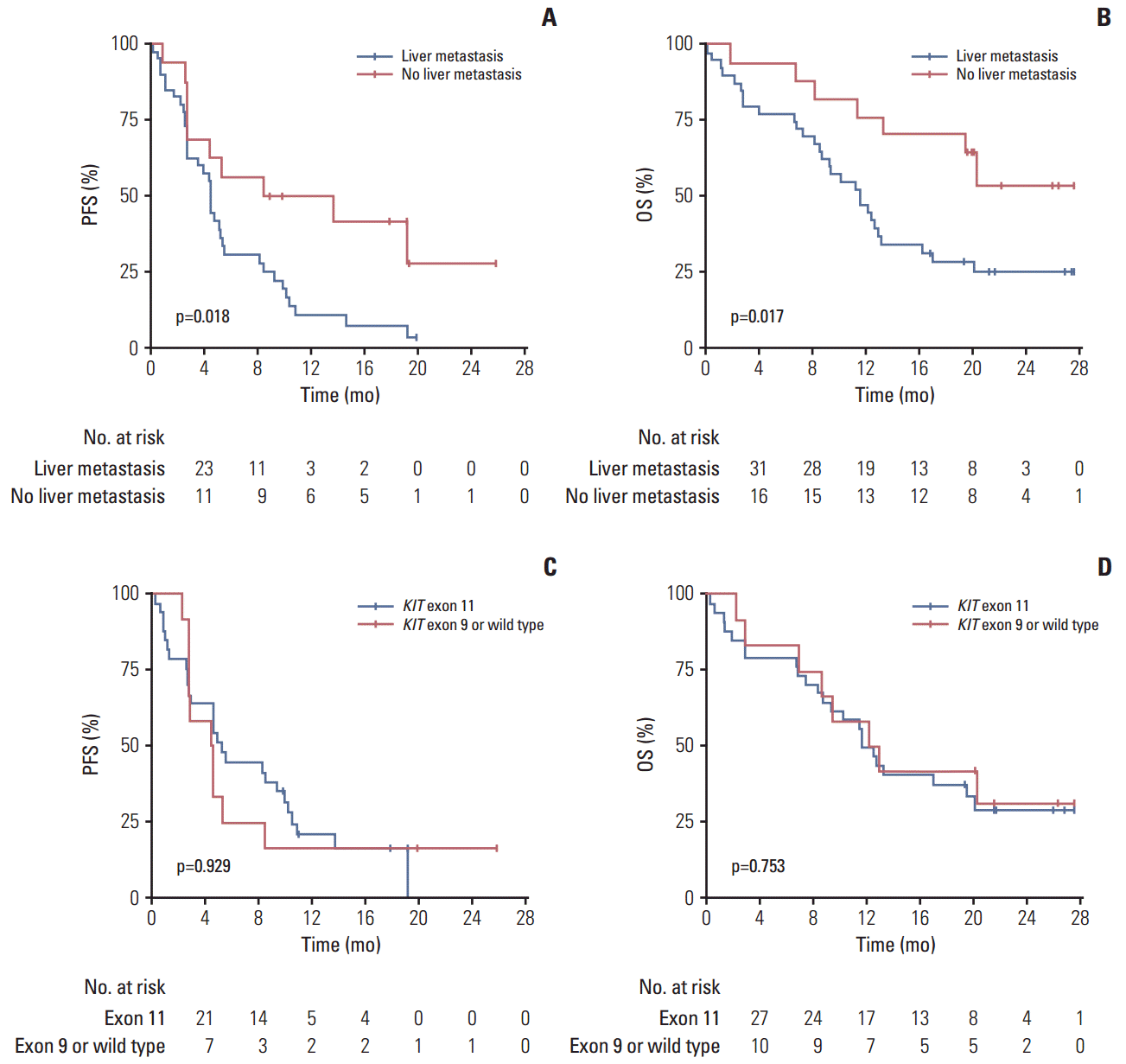
Progression-free survival (PFS) by liver metastasis (A), overall survival (OS) by liver metastasis (B), PFS by primary tumor genotype (C), and OS by primary tumor genotype (D). The p-values were obtained using the log-rank test.

Multivariate analysis for the PFS and OS
3. Safety
During a median of five treatment cycles (range, 1 to 29 cycles), 55 patients (97%) experienced drug-related toxicity (Table 3). Thirty patients (53%) experienced grade 3 adverse events; the most common were HFSR (25%), hypertension (7%), and skin rash (7%). Of these 30 patients with grade 3 adverse events, 21 (70%) experienced a down-grading of their toxicity level to grade 2 or lower, mostly by dose reduction (in 19 patients). An improvement in grade 3 toxicity was not assessable in eight patients (27%, 8/30) because of the short follow-up period after its appearance. No patient experienced grade 4 toxicity, and there were no treatment-related deaths. Regorafenib was dose-reduced in 44 patients (77%) for the following reasons: HFSR (n=31), fatigue (n=5), hypertension (n=4), skin rash (n=4), and vomiting (n=2). The majority of patients (82%, 36/44) underwent a dose reduction within two treatment cycles (after one cycle, 26 patients; after two cycles, 10 patients). Dose reduction was down to 120 mg once daily in 27 patients (61%, 27/44) and to 80 mg once daily in 17 patients (39%, 17/44). The dose was reduced to 80 mg after a median of three treatment cycles (range, 1 to 7 cycles) after an initial dose reduction to 120 mg. After a dose reduction, 33 patients (75%, 33/44) experienced an improvement of their toxicity symptoms. Of these, 29 patients (88%, 29/33) experienced this improvement during the cycle when the regorafenib dose was reduced. Of the 44 patients who required a regorafenib dose reduction, the dose was re-escalated after sustained improvement in the toxicity impacts in 12 patients (27%). This dose re-escalation was performed after a median of two cycles (range, 1 to 7 cycles) had elapsed since a dose reduction. Of these 12 patients, three patients tolerated the escalation, and eight patients required a re-reduction due to a return to the toxicity levels that caused the previous dose reduction in six patients and another toxicity in two patients. A dose escalation from 120 to 160 mg was achieved in eight patients, two of whom (25%) tolerated this higher dose. One of the four patients who received a dose escalation from 80 to 120 mg tolerated this regimen. The tolerability of a dose escalation could not be assessed in one patient because of the short follow-up period.
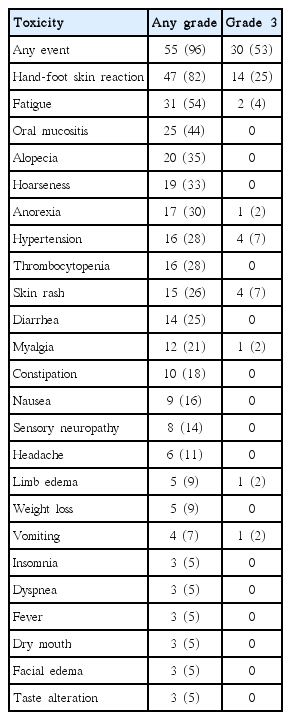
Adverse events occurring in ≥ 5% of patients
4. Rest period symptom exacerbation
Fifteen patients (26%) experienced an exacerbation of their cancer-related symptoms during the rest period in the regorafenib regimen. Symptom exacerbation in the rest period was considered to be present when the symptoms occurred after stopping regorafenib during the rest period and improved with the restarting of regorafenib during the medication period. The exacerbation of these symptoms started after a median of three cycles (range, 1 to 11 cycles) of regorafenib and improved during the regorafenib medication period. The median time to symptom exacerbation during the rest period was the third day of the rest period (range, 2nd to 7th) in the 10 patients, whose onset date of symptom exacerbation was available. The exacerbated symptoms included abdominal pain (n=8), abdominal distension (n=5), palpable abdominal mass (n=2), poor oral intake (n=2), constipation (n=1), fatigue (n=1), and nausea (n=1). Most patients experienced grade 1 (n=6) or grade 2 (n=7) symptoms, and two patients complained of exacerbated grade 3 symptoms. Interventions following the exacerbation of symptoms included the additional medication (n=6), continuation of regorafenib during the rest periods (n=5), and observation (n=4). The additional medications involved pain killers for abdominal pain (n=4), stool softeners for constipation (n=1), and an appetite enhancer for poor oral intake (n=1). The four patients who received pain killers experienced an improvement in their symptoms with these medications, while the two patients on the other medications did not show this improvement. The exacerbation of symptoms was resolved in all five patients given regorafenib during the rest period. The exacerbated symptoms in these five patients were abdominal pain (n=3), abdominal distension (n=2), extremity edema (n=1), palpable abdominal mass (n=1), and poor oral intake (n=1). The regorafenib dose taken in the rest period was the same as that taken in the medication period. Two patients continued regorafenib in the rest period due to an exacerbation of abdominal pain despite the increase in pain killers. One patient complained of an exacerbation of HFSR after a continuation of the regorafenib during the rest periods, which were improved by a dose reduction. The other four patients did not complain of any exacerbation of their treatment-related symptoms after the continuation of regorafenib during the rest period. All four patients in the observation scheme continued to complain of an exacerbation of their symptoms during the rest period.
Discussion
Regorafenib has shown similar clinical efficacy to that of the GRID trial in Korean patients with advanced GIST who experienced treatment failure at least with imatinib and sunitinib. In accordance with previous studies [11,12], this clinical benefit in the present study population was attributed mainly to disease control rather than tumor reduction. After failure with imatinib and sunitinib, the patients with an advanced GIST have few therapeutic options and can generally be treated with a palliative aim. The resumption of imatinib can be one option and has shown a longer PFS (1.8 months; HR, 0.48; 95% CI, 0.28 to 0.82) than that in the placebo groups (0.9 months) in patients with advanced GIST after failure with imatinib and sunitinib (RIGHT study) [14]. Regorafenib has shown a longer PFS (4.5 months in this study and 4.8 months in GRID trial) than that in the RIGHT study imatinib retrial group (1.8 months) and in the placebo groups in the RIGHT or GRID trial (0.9 months) [12,14]. The role of regorafenib was supported further by recent analysis that suggested a survival benefit for regorafenib in GIST with a correction for the impact of cross-over in the GRID trial [15]. The median OS was reported to be 17.4 months in this updated OS analysis of the GRID phase III trial [15], and 12.9 months in the current study. Furthermore, one study suggested that the quality of life was similar in patients with advanced GIST in both the regorafenib and placebo groups [16].
The present study population had similar treatment-related toxicity profiles to those of the previous phase II and phase III GRID trials but a lower incidence of hypertension and diarrhea (49% and 40% in GRID trial vs. 28% and 25% in this study) [11,12]. HFSR was the toxicity observed most frequently and the most common reason for dose reduction in the present study. The incidence of HFSR in the study population (82%) was higher than that in the regorafenib group in the GRID trial (56%) [12] but was similar to that in the regorafenib group in the GRID trial Japanese subgroup (92%) [13]. The toxicity was managed successfully with a dose reduction. Therefore, only four patients discontinued regorafenib due to the drug-related adverse events.
The exacerbation of cancer-related symptoms during the rest period was troublesome in approximately 25% of the study patients. A continuation of regorafenib during the rest period was an effective way of improving these symptoms. All five patients who received a continuation of regorafenib during the rest period experienced an improvement in their symptoms, and only one of these cases complained of a temporary exacerbation of their drug-related toxicity. Although in most cases, pain killers were found to be effective in controlling the exacerbation of abdominal pain during the rest period, a continuation of regorafenib through a rest period can reduce the level of abdominal pain when pain killers are ineffective. A similar case of a patient with rectal GIST, who complained of pelvic pain during a 1-week break period of regorafenib and then experienced an improvement of their pain with continuously administered regorafenib, was reported recently in Japan [17]. Additional medications were not found to be effective in controlling the exacerbation of their cancer-related symptoms during the rest period. Furthermore, symptoms, such as abdominal distension and palpable abdominal masses, were believed to be related to tumor progression. In addition, symptom control by the continuation of regorafenib during the rest period suggests that tumor progression can occur during the rest period and a continuous dosing schedule can prevent disease exacerbation during the rest period with an intermittent dosing schedule. Tumor cell regrowth induced by the removal of regorafenib [18] as well as the clinical efficacy and tolerability of once-daily continuous dosing of regorafenib [19] were reported in other malignancies. A future study will evaluate the continuous regorafenib regimens in the patients with GIST will strengthen these findings.
Conclusion
Regorafenib has shown similar clinical efficacy to that in the GRID trial in Korean patients with advanced GISTs after a failure of imatinib and sunitinib. The toxicity could be controlled successfully with a dose reduction in these cases. The exacerbation of their cancer-related symptoms during the rest periods is manageable with the continuation of regorafenib.
Notes
Conflict of interest relevant to this article was not reported.