Introduction
As we currently live in an era of information and advanced genomics, we should maintain our focus on how we respond to and process the overwhelming amount of information we encounter. For example, Mandel and Metais [1] first described the presence of nucleic acids in human blood in 1948, but several decades passed before attention was paid to the vast amount of information supplied by nucleic acids in the blood. However, since the discovery of mutant RAS gene fragments in the blood of cancer patients in 1994 [2] and the detection of microsatellite DNA changes in the serum of cancer patients in 1996 [3], the information contained within the nucleic acids in the blood has gradually gained attention.
Blood contains cellular components and numerous biological substances, such as extracellular vesicles, proteins, and nucleic acids, including mRNAs, miRNAs, and cell-free DNA (cfDNA). cfDNA refers to any non-encapsulated DNA within the bloodstream originating from various cell types. The portion of the cfDNA in the blood of cancer patients released from tumor cells via apoptosis, necrosis, or active release [4,5], is commonly referred to as the circulating tumor DNA (ctDNA). The ctDNA has gained increasing attention since 2010 because of the potential to detect early cancer metastases through novel, sensitive laboratory methods that cannot be detected by high-resolution imaging techniques [6]. The BRACAnalysis (Myriad Genetic Laboratories, Salt Lake City, UT) was the first Food and Drug Administration (FDA) approved companion diagnostic test using ovarian cancer patient’s blood specimens with the development of a gene mutation treatment. Expectations arose that ctDNA could lead to drug treatments for cancer patients. Since then, the number of tests and studies related to ctDNA has exploded exponentially (Fig. 1).
Nucleic acids in the blood are heterogenous depending on their origin. ctDNA analysis can provide more comprehensive information than a conventional tissue biopsy, which has the spatial limitation inherent in sampling due to tumor tissue heterogeneity. It is estimated that up to 3.3% of tumor DNA enters the blood daily from 100 g of tumor tissue, equivalent to 3×1010 tumor cells [7]. On average, the size of the ctDNA varies from small fragments of 70–200 base pairs to large fragments of up to 21 kb [8]. It is important to note the relatively short half-life of ctDNA in blood circulation, ranging from 16 minutes to 2.5 hours [9,10]. Although many tumor-specific abnormalities (e.g., mutations in tumor or tumor suppressor genes, changes in DNA integrity [11], abnormal gene methylation, changes in microsatellite [3], mitochondrial DNA loading levels, and changes in chromosomal genomes [12]) can be detected using ctDNA, a number of obstacles exist in the implementation of ctDNA for screening and diagnosis. First, normal hematopoietic cells and other nucleic acids of non-tumor origin also contribute to the ctDNA in the blood and cause false positives in ctDNA assays in cancer patients [13,14]. Not all somatic mutations detected in the ctDNA analyses are of cancer origin; clonal expansion of somatic variants can be observed in healthy individuals and may represent clonal hematopoiesis of indeterminate potential (CHIP). CHIP frequency increases with age, with only 1% of people under the age of 50 but > 10% over the age of 65 exhibiting CHIP [15–17]. These abnormalities commonly occur in the DNMT3A, TET2, and ASXL1 genes [16], but have also been reported in other genes such as TP53, JAK2, SF3B1, GNB1, PPM1D, GNAS, and BCORL1 [15]. Simultaneous occurrence of CHIP and tumor-derived gene mutations that have abnormalities in these genes may cause difficulties interpreting the ctDNA assays. The second issue is the low concentration of ctDNA (1–10 ng/mL in asymptomatic individuals) [18]. Depending on the concentration of ctDNA, a false negative result is possible; therefore, the sample volume is an important factor affecting the results. Third, the variant allele frequency (VAF) of ctDNA is usually much lower, often below 1%, and can be affected by factors such as cancer type, stage, and clearance rate [19]. Any interpretation of the results requires careful decisions regarding the threshold of allele frequencies of the detected variants, as these are critical aspects. Fourth, there is a lack of consensus on how ctDNA detection should be performed, from the extraction stage to the final in silico variant analysis stage. Even the nomenclatures related to ctDNA lack a proper consensus [20]. Due to the rapid incremental clinical use of ctDNA testing, the unmet demand for a proper consensus on ctDNA-related issues remains [20,21].
This review describes the currently-available ctDNA assays based on the different methodologies, ranging from the traditional methods to more recent advanced molecular technologies. We focus on the unmet need for clinical validation of ctDNA testing by reviewing the validation and approval processes of the FDA and European Commission in vitro Diagnostic Medical Device (CE-IVD), among others. This review addresses frequently raised questions regarding the clinical application of ctDNA assays, summarizes the current status of approved and validated ctDNA assays, and the future direction of ctDNA testing.
The A to Z of the ctDNA Test
Before introducing the ctDNA test, the terminology and definitions of ctDNA must be clarified. Bronkhorst et al. [22] proposed a nomenclature system for three highly investigated diagnostic areas based on the biological compartment in which the cfDNA is distributed (depending on its presence in circulation) and the origin (Fig. 2) [23]. cfDNA is highly heterologous, and a broader concept is needed that covers both nuclear and microbial DNA. Nuclear DNA includes mitochondrial DNA, and microbial DNA encompasses both microbial and viral DNA, not of human origin. Part of the nuclear DNA in the plasma of cancer patients is ctDNA. ctDNA usually refers to all types of tumor-derived DNA in the circulating blood, as discussed in this review.
DNA abnormalities occur by different parts, and each has different features. These features of the ctDNA have different potential clinical implications (Fig. 3). Genomic aberrations of somatic origin detectable in the ctDNA include mutations, chromosomal rearrangements, and copy number changes. An additional features characteristic of ctDNA is specific epigenetic aberrations such as methylation patterns or different DNA fragment lengths [24]. Although ctDNA tests vary in their genomic features and coverage of the genes of interest, the basic principles of the test remain the same. Two categories exist; targeted approaches that test for a small number of known mutations, and untargeted approaches that broadly test for unknown targets. A targeted approach includes real-time polymerase chain reaction (RT-PCR), digital PCR (dPCR), and beads, emulsion, amplification, and magnetics (BEAMing) technology, whereas the broader approach would include high-throughput sequencing methods based on next-generation sequencing (NGS), whole exome sequencing (WES), whole genome sequencing (WGS), and mass-spectrometry-based detection of PCR amplicons [25] among others.
1. ctDNA detection methods
1) RT-PCR
RT-PCR is widely used for variant screening because it is relatively inexpensive and fast [26]. The variants are detected via the binding of complementary sequences using fluorescent-labeled sequence-specific probes, and the fluorescence intensity is related to the amount of amplified product. The sensitivity of RT-PCR is approximately 10%, which is lower than that of other test methods [27,28]. Cold amplification at a lower denaturation temperature PCR (COLD-PCR) is a variant assay that improves the RT-PCR sensitivity. COLD-PCR concentrates mutated DNA sequences in preference to the wild type using a lower-temperature denaturation step during the cycling protocol. The denaturation temperature for a given sequence is adjusted within ±0.3°C to allow for selective denaturation and amplification of mutated sequences, while double-stranded wild-type sequences are amplified less. This assay can enrich the mutant sequences, improving the sensitivity to detect the mutant allele frequency (MAF) to approximately 0.1% [29,30]. The PCR-based method has the advantage of high sensitivity and cost-effectiveness but is limited as only known variants can be selected with limiting input and speed.
2) Digital PCR
dPCR shares the same reaction principle as RT-PCR, except that the samples are dispersed into arrays or droplets, resulting in thousands of parallel PCR reactions. The dPCR can quantify a low fraction of variants against a high background wild-type cfDNA using a single or few DNA templates in an array/droplet and has 0.1% sensitivity [26]. The dPCR method can be applied to cancer personalized profiling by deep sequencing (CAPP-Seq) in combination with molecular barcoding technologies that improve sensitivity by reducing the background sequencing error [31,32]. These two methods improve the sensitivity of CAPP-Seq up to three-fold and, when combined with molecular barcoding, yield approximately 15-fold improvements [32]. cfDNA enrichment is conducted by a two-step PCR procedure during the sample preparation process. The first PCR amplifies the mutational hotspot regions of several genes in a single tube. The second PCR is a nested PCR with unique barcoded primers for sample labeling. The final PCR products are pooled and partitioned for sequencing. The advanced dPCR assay (BEAMing) is a highly sensitive approach with a detection rate of 0.02% [33,34]. This approach consists of four principal components: beads, emulsion, amplification, and magnetics. BEAMing combines dPCR with magnetic bead and flow cytometry [27,33]. In BEAMing, the primer binds to the magnetic beads using a reaction that forms a biotin-streptavidin complex. Less than one template molecule and less than one bead are contained within the microemulsion, and the PCR is performed within each droplet. At the end of the PCR process, the beads are magnetically purified. After denaturation, the beads are incubated with oligonucleotides to distinguish between different templates. The bound hybridization probe is then labeled with a fluorescently labeled antibody. Finally, the amplified products are counted as fluorescent beads by flow cytometry. However, the BEAMing method is impractical for routine clinical use due to its workflow complexity and high cost [34,35].
3) Mass spectrometry
The mass spectrometry-based method combines the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry with a conventional multiplex PCR. An example of this method is UltraSEEK (Agena Bioscience, San Diego, CA). UltraSEEK consists of two-step PCR for amplification and mass spectrometry for detection. The two-step PCR step consists of a multiplex PCR followed by a mutation-specific single-base extension reaction. The extension reaction uses a single mutation-specific chain terminator labeled with a moiety for solid phase capture. Captured, washed, and eluted products are examined for mass, and mutational genotypes are identified and characterized using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry [36]. UltraSEEK has the advantage of multiplex detection of mutant sequences simultaneously, and has a MAF of 0.1% [37].
4) Next-generation sequencing
NGS, also known as massively parallel sequencing technology, can characterize cancer at the genomic, transcriptomic, and epigenetic levels. NGS is a highly sensitive assay that can detect mutations in MAF of < 1% using the latest platforms [38]. NGS can analyze several million short DNA sequences in parallel and conduct sequence alignment or de novo sequence assembly to the reference genome [39]. Depending on the panel configuration, NGS panels can be targeted to analyze known variants or untargeted to screen unknown variants. Target panels were preferred due to their high sensitivity and low cost but are limited to point mutations and indel analysis. Several NGS methods can be applied to target panels with adjustable sensitivity, including tagged amplicon deep sequencing, the safe sequencing system, and CAPP-Seq. On the other hand, WGS or WES using untargeted panels allow detection of unknown DNA variants throughout the entire genome (or exome). Different genome-wide sequencing methods have been proposed for different variation types, such as personalized analysis of rearranged ends, digital karyotyping, and Fast Aneuploidy Screening Test-Sequencing System [35]. However, genome-wide sequencing requires a large sample, making its application for ctDNA difficult due to the low concentrations of ctDNA in samples.
There have been attempts to analyze DNA fragmentation differences. There is a marked difference in fragment length size between ctDNA and normal cf DNA. The fragment length of ctDNA is consistently shorter than that of normal cfDNA [40]. Besides, ctDNA with a low MAF (< 0.6%) is associated with a longer ctDNA fragment length when compared to normal cfDNAs [41]. Moreover, most cancers of different origins showed fragmentation profiles of varying lengths [42]. The characteristic DNA fragmentation provides a proof-of-principle approach applicable to screening, early detection, and monitoring of various cancer types.
NGS application has been extended to microsatellite inst-ability (MSI) detection [43]. Loss of DNA mismatch repair (MMR) activity leads to an accumulation of mutations that could otherwise be corrected by MMR genes. A deficiency in MMR activity is often caused by germline mutations or aberrant methylation. The MSI phenotype of a deficiency in MMR activity refers to the shortening or lengthening of tandem DNA repeats in coding and noncoding regions throughout the genome. Tumors with at least 30% to 40% of unstable microsatellite loci, termed microsatellite instability high (MSI-H) [43], reportedly have a better prognosis than MSS tumors and tumors with low MSI. MSI has been documented in various cancer types, including colon, endometrium, and stomach cancers [44,45]. The FDA has approved Pembrolizumab to treat MSI-H cancer regardless of the tumor type or site [46]. NGS-based methods utilize various MSI detection algorithms such as MSIsensor [47], mSINGS [48], MANTIS [49], and bMSISEA [43], which have demonstrated concordance rates ranging from 92.3% to 100% with the PCR-based method. The application of NGS can reliably detect the MSI status with a ctDNA fraction up to 0.4% [43].
5) Methylation analysis
Epigenetic information such as methylation is more specific to the tissue of origin than genetic mutations [50]. Changes in DNA methylation patterns occur early in tumor development and have been reported to help early screening for cancers of unknown origin [35,51]. Methylation analysis is not routinely or commonly used to detect ctDNA, but it can be partially applied to cancer patients. The method can be broadly divided depending on its application to the candidate gene. The Grail’s technology applied DNA methylation patterns to differentiate between cancer cell types or tissue origins [52]. Most cfDNA methylation analysis methods applied a candidate gene approach due to the low analytical cost and the efficiency of using pre-established epigenetic biomarkers [53]. Bisulfite treatment-based assays distinguish cytosine methylation and are generally the preferred ctDNA methylation detection method [54]. The analytical principle is based on treating the DNA with bisulfite to convert unmethylated cytosine residues to uracil. Two types of ctDNA methylation analysis exist; PCR-based methods that apply specific primers or melting temperatures and sequence-based methods such as direct sequencing or pyrosequencing. However, the accuracy of bisulfite pyrosequencing is only maintained up to 5% [55,56]. Methylation-specific PCR (MSP) can distinguish DNA sequences by sequence-specific PCR primers after bisulfite conversion [57]. The methylation-sensitive high-resolution melting (MS-HRM) protocol is based on comparing the melting profiles of the PCR products from unknown samples with profiles for specific PCR products derived from methylated and unmethylated control DNAs [58]. The protocol consists of PCR amplification of bisulfite-modified DNA with primers and subsequent high-resolution melting analysis of the PCR product. MSP or MS-HRM can accurately detect about 0.1% of methylated DNA [57,58].
6) Hybrid sequencing (NanoString)
The nCounter Technology (NanoString Technologies, Seattle, WA) is a novel technology developed to screen clinically-relevant ALK, ROS1, and RET fusion genes in lung cancer tissue samples. NanoString is applicable to RNA, miRNA, or protein and, more recently, to ctDNA [59–61]. Target ctDNA is directly tagged with capture and reporter probes that are specific to the target variant of interest, creating a unique target-probe complex. The probes include a fluorescent reporter and a secondary biotinylated capture probe that allows immobilization onto the cartridge surface. The target-probe complex is immobilized and aligned on the imaging surface. The labeled barcode of the complex is then directly counted by an automated fluorescence microscope [62,63].
2. International efforts for advanced precision medicine in ctDNA analysis
The availability of new ctDNA testing methods and continuous scientific advances has resulted in several new problems. The factors affecting ctDNA testing outcomes are present from the sample collection phase to the final reporting phase. The American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) reviewed the framework for future research into clinical ctDNA tests in 2018 [64]. This article categorizes the key findings that affect ctDNA testing for oncology patients into preanalytical variables for ctDNA specimens, analytical validity, interpretation and reporting, and clinical validity and utility of each test. Plasma is the most suitable sample recommended for ctDNA testing [65], as is the use of specific types of sample collection tubes such as cell-stabilizing tubes (Cell-Free DNABCT [STRECK tubes] and PAXgene Blood DNA tubes [Qiagen]), or conventional EDTA anticoagulant tubes [66–68]. Leukocyte stabilization tubes can extend the preprocessing window to 48 hours after collection, but EDTA anticoagulant tubes require processing within 6 hours. However, few studies have examined the preanalytical variables affecting ctDNA testing, and guidelines are needed to validate their clinical utility. Considering the variations in the many factors and different types of ctDNA assays, based on different methods, the validity of each analysis must be comparable. The current clinical ctDNA analyses require a clear assessment of the validity of the individual analyses. To increase the precision of ctDNA assays, best practices, protocols, and quality metrics for NGS-based ctDNA analyses must be developed.
The Sequencing Quality Control Phase 2 (SEQC2) consortium organized by the FDA is an international group of members from academia, government, and industry (https://www.fda.gov/science-research/bioinformatics-tools/microarraysequencing-quality-control-maqcseqc#MAQC_IV). The SEQC2 Oncopanel Sequencing Working Group developed a translational scientific infrastructure to be applied for practices in precision oncology [69]. The Oncopanel Sequencing Working Group evaluated panels/assays, genomic regions, coverage, VAF ranges, and bioinformatics pipelines, using self-constructed reference samples. This study on the analytical performance evaluation of oncopanels/assays for small variant detection includes: (1) comprehensive solid tumor oncopanel examination [70], (2) liquid biopsy testing [71], (3) testing involving formalin-fixed paraffin-embedded material [72], and (4) testing involving spike-in materials [73]. A major finding from the SEQC2 liquid biopsy proficiency testing study [71] is that all assays could detect mutations with high sensitivity, precision, and reproducibility for those above the 0.5% VAF threshold. The degree of DNA input material impacted the test sensitivity, requiring higher input for improved sensitivity and reproducibility for variants with a VAF below 0.5%.
Advanced NGS-based assays for precision oncology are in high demand, and recently approved ctDNA assays are to be identified. Establishing a proper validation scheme would support the FDA’s regulatory and scientific endeavors.
3. FDA-approved ctDNA assay
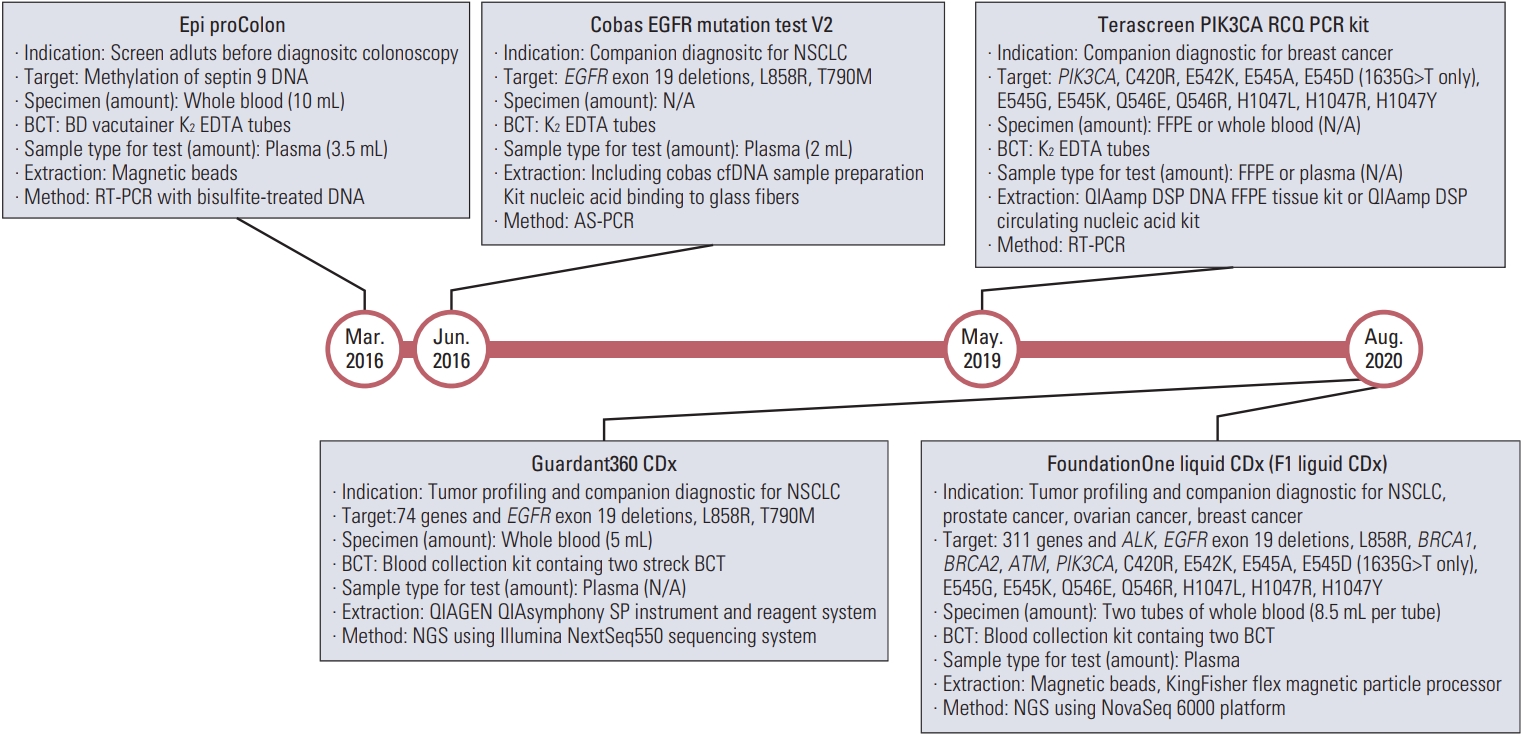
We searched for FDA-approved assays in the FDA database (https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm) using the following keywords: circulating tumor DNA, ctDNA, cell-free DNA, circulating cell-free DNA, cfDNA, liquid biopsy, and plasma and DNA. The search results were compared to the annual report of medical devices cleared or approved on FDA lists published between 2013 and 2022 for confirmation, and assays related to ctDNA were selected. We identified three in vitro diagnostic devices (Epi ProColon, Cobas EGFR Mutation Test, and therascreen PIK3CA RGQ PCR Kit) and two specialized laboratory services (Guardant360 CDx, and FoundationOne Liquid CDx) (Fig. 4).
FoundationOne Liquid CDx was approved as a companion diagnostic on October 26 and November 6, 2020. The approved companion diagnostic indications are (1) to identify mutations in BRCA1 and BRCA2 genes in patients with ovarian cancer eligible for treatment with rucaparib (RUBRACA, Clovis Oncology, Inc.), (2) to identify ALK rearrangements in patients with non–small cell lung cancer eligible for treatment with alectinib (ALECENSA, Genentech USA Inc.), (3) to identify mutations in the PIK3CA gene in patients with breast cancer eligible for treatment with alpelisib (PIQRAY, Novartis Pharmaceutical Corporation), and (4) to identify mutations in the BRCA1, BRCA2, and ATM genes in patients with metastatic castration-resistant prostate cancer eligible for treatment with olaparib (LYNPARZA, AstraZeneca Pharmaceuticals LP) [74]. The NGS-based ctDNA tests related to companion diagnostics, such as FoundationOne Liquid CDx and Guardant360 CDx are transitioning to specialized laboratory services. FDA-approved tests require evaluations of their analytical performance. Recent laboratory-based tests have undergone extensive evaluation testing using large sample numbers for advanced assay interpretation and reporting, clinical validation, and utility (Table 1). Considering the cost of the tests, the number of tests performed for evaluation is prohibitive for small laboratories. Specialized laboratories use their own processes (Table 2) to provide users with reports. Therefore, testing is changing from a complex assay performed at individual laboratories to a more specialized service where each specialized laboratory be devised its own analysis processes, and the type of inspection changes to a laboratory service.
4. CE-marked ctDNA assay
In May 2017, the Conformité Européenne (CE) declared the strengthening of in vitro Diagnostic Regulation and Medical Device Regulation regulations. The transition was completed in May 2022, following a 5-year transition period. The CE announced a new database search service called the European Database on Medical Devices (EUDAMED), which will consist of six modules: actor registration, unique device ID and device registration, notification authority and certificate, clinical and performance research, and alert and market monitoring. The EUDAMED database was scheduled for release in July 2022 but has been postponed to the third quarter of 2024 due to a delay in the module development process. We had difficulty performing a systematic search for medical devices or in vitro diagnostics with the CE mark. Therefore, we searched for recently published papers that mentioned CE-marked products (Table 3).
5. Marketplace of ctDNA test in Republic of Korea
In Korea, the marketplace of ctDNA test has recently expanded as the In Vitro Diagnostic Medical Devices Act was promulgated in April 2019. When ctDNA test was searched in the medical device database of the Korea Ministry of Food and Drug Safety (https://udiportal.mfds.go.kr/search/data/P02_01#list), a total of seven domestic tests were identified. The Smart Biopsy EML4-ALK Detection Kit (CytoGen, Seoul, Korea) was first nationally accredited on April 20, 2016, and the following tests have been nationally accredited in sequence; ADPS EGFR Mutation Test Kit V1 (GENECAST, Seoul, Korea), Droplex KRAS Mutation Test v2 (Gencurix, Seoul, Korea), Droplex PIK3CA Mutation Test (Gencurix), PANAMutyper R EGFR V2 (PANAGENE, Daejeon, Korea), AlphaLiquid 100 (IMBDX, Seoul, Korea), and LiquidSCAN (GENINUS, Seoul, Korea). Most tests are PCR-based assay, such as RT-PCR and dPCR, but AlphaLiquid 100 (IMBDX) and LiquidSCAN (GENINUS) are NGS-based assay.
Current and Future Directions
The introduction of ctDNA testing and technical advancements in NGS have affected cancer’s diagnostic and therapeutic aspects. Many biomarkers associated with treatment options have been identified for cancer patients whose tissues were previously unavailable for biopsies. The widespread use of NGS and its increased availability has changed the concept/scheme of companion diagnostics from ‘one gene-one drug’ to ‘multi-genes - multi-drugs’ treatment [75]. Recently, experts from the National Comprehensive Cancer Network have recommended measuring multiple predictive genes associated with companion diagnostics for certain cancers [76].
Although tissue biopsy remains the standard of diagnosis because of its important pathological diagnostic information value and the need to assess biomarkers without DNA alterations, such as estrogen receptor expression and other protein or RNA biomarkers [77]. However, ctDNA testing is undoubtedly a very promising technology, with broad clinical applications for early diagnosis, monitoring, management, and prognosis [78,79]. When performing metastatic diagnosis alongside standard tissue biopsies, ctDNA testing can provide key advantages, either as a baseline for follow-up testing after treatment or in situations in which more-rapid identification of targetable alterations is needed to guide first-line therapy [77]. In addition, ctDNA testing plays an important role in real-time monitoring of various aspects of tumors due to its simple sample preparation [78].
We expect that the strengths of ctDNA, including the potential ability to detect latent cancers and track tumor-specific mutations, will naturally enable minimal residual disease (MRD) assessment [80]. The ability to identify microscopic residuals and occult metastases could revolutionize the individualization of adjuvant and consolidation therapy [81]. Despite the potential use of ctDNA to determine MRD, it is premature for use in this feature due to many current issues [82,83]. Therefore, the reliability and clinical validity of ctDNA analysis is becoming increasingly important as it can directly impact patient care with respect to treatment options.
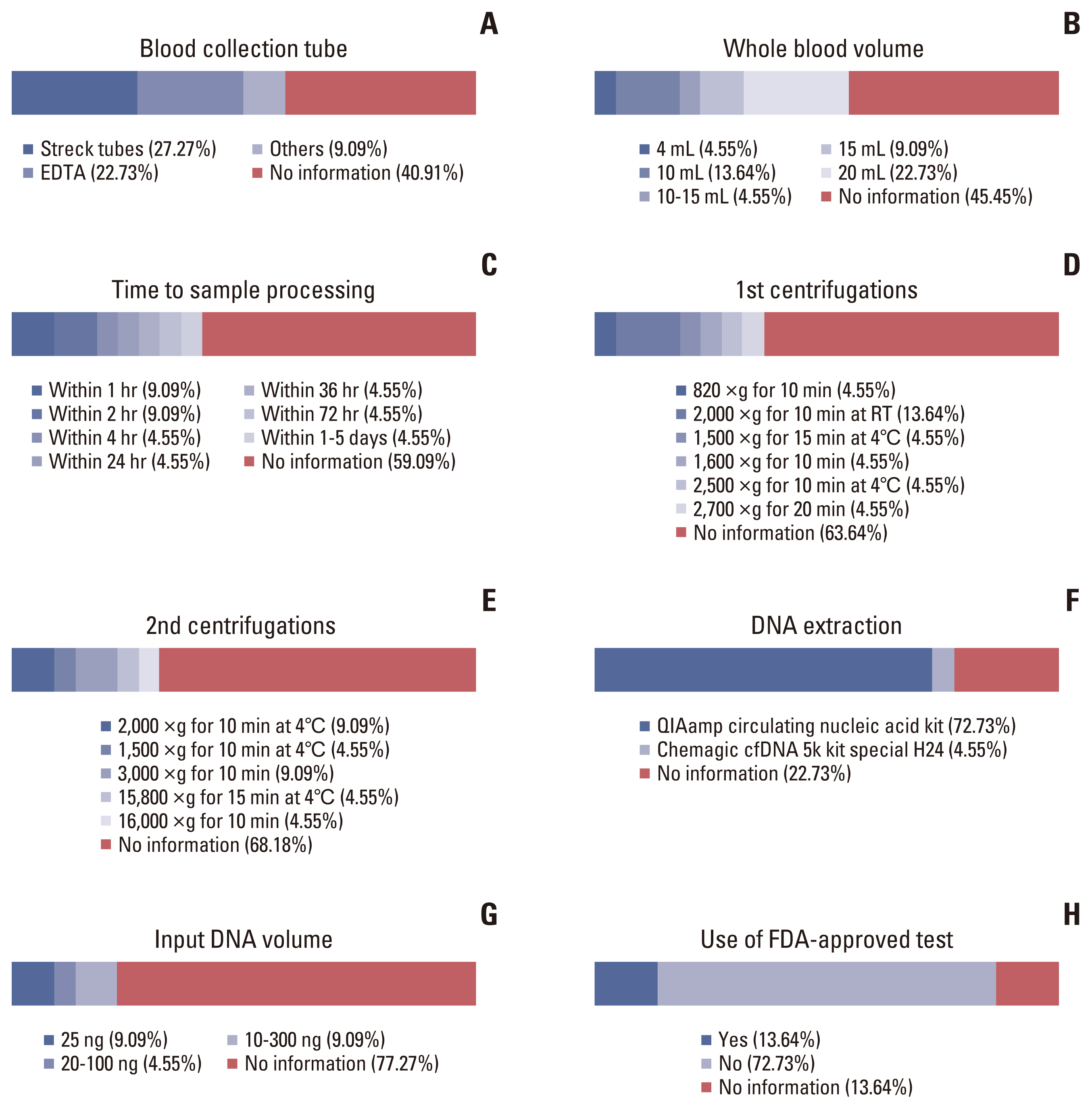
To assess the current status of ctDNA testing and ongoing developments, we searched the clinical trial database of the FDA (Clinical trial.gov). A query using ctDNA as the keyword showed 978 clinical trials as of June 2022. The results of 109 trials in which the clinical trials were completed were reviewed using the uploaded articles from the database or the national clinical trial number for a PubMed search. Twenty-two clinical trials were available, and we reviewed and compared their preanalytical and analytical variables (Fig. 5). The preanalytical variables of the blood collection tube used, the volume of whole blood collected, time to sample processing, centrifugation protocols, and DNA extraction methods were missing or unidentifiable in over half of the reports, despite their importance. When provided, the information varied among studies; specimen processing within 24 hours using EDTA tubes was a possible confounding factor regarding the stability of the ctDNA. Some trials requiring detection of low VAF variants, such as ‘using copy number variation of ctDNA for cancer diagnosis’ or ‘biomarker response according to treatment in metastatic cancer’ used only 25 ng DNA, which appears to be insufficient and, therefore, they were unable to exclude the possibility of false negatives. The use of FDA-approved assays among trials was low at 13.64% (3/22). Information regarding approval from other institutions or agencies was often unavailable, but most clinical trials (72.73%, 16/22) utilized non–FDA-approved testing methods.
Despite the considerable therapeutic influence of ctDNA testing or companion diagnostics, the current practice of utilizing various ctDNA tests without regard to a consensus on clinical validation is questionable, as the review of clinical trials and available information demonstrates. Discrepant test results between the tissue biopsy and ctDNA results are common, and the underlying reasons for these discrepancies include temporal heterogeneity (an archival tumor specimen), spatial heterogeneity (a subclonal mutation), and analytical errors [64]. In the case of analytical errors, the source of the error should be evaluated before any therapeutic action can be taken. If such an investigation or validation is lacking, this should be disclosed to enable the participants or patients to give proper informed consent. The common rules followed by institutional review boards (IRB) when reviewing research state that the prospective participants (or a legally authorized representative) be provided with sufficient detailed information regarding the research. The consent form containing this information must be organized to facilitate an understanding of why one might or might not want to participate [84,85]. This should also be the case if the patient opts for a ctDNA test. Patients should be able to choose the ctDNA test based on detailed information about the accuracy of the test, the list of genes that can be analyzed, and the laboratory’s ability to analyze mutations based on experience.
Laboratories must be aware of any new developments in ctDNA testing. A changing trend in ctDNA testing is demonstrated by the recent FDA-approved ctDNA assays. Previously FDA-approved assays were mostly in vitro diagnostic devices (IVDs), conducted by small-scale clinical laboratories. However, recent FDA-approved assays require referrals to larger, specialized laboratories with institutional accreditation. Such changes are inevitable due to the testing complexity and higher reliability required by clinical practice. Cutting-edge ctDNA testing is costly and requires first-rate laboratory infrastructure and highly specialized and multi-disciplinary professionals. The trend toward centralization and referrals is in line with these requirements.
Previously, the introduction of tumor markers has resulted in the overutilization of tumor marker testing in the hope of providing definitive answers to cancer diagnostics. The need for specific guidelines/instructions on how tumor markers should be utilized demonstrates the concern regarding the misuse of tumor markers, potentially resulting in misdiagnosis or a delay in treatment [86]. In recognition of these issues, in 2002, the National Academy of Clinical Biochemistry produced the Laboratory Medicine Practice Guideline of tumor biomarkers [87]. The guideline provides recommendations based on expert opinions from those in the field of IVD and the marketplace. This regulatory guideline includes 16 different cancers and their established tumor markers, their qualities, and the technological requirements. It is anticipated that a similar development to the guideline for tumor marker testing will be available for ctDNA testing soon. However, guidelines on validation are currently lacking. ctDNA testing requires clinical validation prior to its clinical implementation. These regulated clinical validation guidelines will inevitably require updating, refinement, and modification as knowledge and understanding of ctDNA and its biological role increases.
In summary, ctDNA testing requires a minimum safety resolution through clinical validation to ensure its clinical utility. The testing requires cooperation between multi-disciplinary experts to provide meaningful and reliable results. Establishing a proper clinical validation guideline for ctDNA will enable access to better cancer treatment and reliable testing in the future.