Introduction
Diffuse intrinsic pontine glioma (DIPG) accounts for 10%–20% of all childhood brain tumors, and has a poor prognosis with a median overall survival (OS) of less than 12 months [1–3]. Approximately 10% of pediatric patients with DIPG survive for ≥ 2 years after diagnosis [4]. Based on recent advances in molecular profiling, the World Health Organization (WHO) defined a new pathologic entity, “diffuse midline glioma, H3K27M-mutant” [5]. The pathologic diagnosis of this new variant is defined by the presence of a somatic mutation at position K27 in one of the several histone-encoding genes. DIPG cases with H3K27M mutation, which account for approximately 80% of clinically recognized DIPGs, have a significantly worse prognosis than those without the H3K27M mutation [6]; however, to date, there is no known treatment for such cases in clinical practice.
Owing to the anatomical complexity and critical functions of the brainstem, surgical resection of DIPG is rarely performed, and pathologic diagnosis via biopsy is very limited. Radiotherapy remains the standard of care, delaying the progression of disease for some months [7]. However, radiotherapy techniques, such as optimal target volume, radiotherapy modality, and dose schemes, are not well established. To improve prognosis, several studies have investigated the additive use of chemotherapy and radiotherapy with inconclusive findings [8,9]. Bevacizumab, an anti-vascular endothelial growth factor antibody, was investigated as a novel therapeutic agent; however, its efficacy in children with DIPG remains unclear. Despite medical progress, overall research on DIPG remains scarce due to tumor rarity, unclear pathology, and ineffective therapies. This multicenter retrospective study aimed to investigate prognostic factors associated with survival in children newly diagnosed with DIPG and treated with radiotherapy in Korea.
Materials and Methods
1. Study population
Medical records of patients with newly diagnosed DIPG treated with radiotherapy between January 2000 and December 2018 at 10 participating centers were retrospectively reviewed based on the Korean Radiation Oncology Group (KROG) 20-01 protocol.
Patients aged < 30 years who had undergone gadolinium-enhanced magnetic resonance imaging (MRI) at diagnosis, presenting with radiologic features consistent with DIPG, were included in the study. DIPG was diagnosed when MRI showed a T1-hypo (or iso) intense and T2-hyperintense lesion involving at least 50% of the pons. Patients with focal brainstem glioma, dorsally exophytic tumor, and extrinsic tumors secondarily invading the pons were excluded. Moreover, patients who were pathologically diagnosed with a WHO grade 1 glioma or non-glioma histology and those whose follow-up data could not be obtained were also excluded. All included patients had been treated with radiotherapy. Histological diagnosis through stereotactic biopsy or resection was not essential, as DIPG is routinely diagnosed based on radiologic findings.
2. Clinical and radiologic variables
Data on clinical characteristics, including age, sex, Karnofsky/Lansky (for patients aged < 16 years) performance status, symptoms, and symptom duration were extracted from the patients’ medical records. Symptoms were categorized as: cranial nerve palsy, cerebellar signs (nystagmus, dysarthria, dysmetria, or ataxia), and pyramidal tract signs (mono-, hemi-, or quadriparesis; hyperreflexia; or positive Babinski sign). Histopathologic data were obtained for patients who underwent biopsy or surgical resection. Data on molecular markers, including H3K27M mutation, O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, IDH1 mutation, p53, Ki-67, and 1p19q co-deletion, were collected, as available.
Radiologic findings of the tumor were assessed by neuro-radiologists from each institution for factors such as extrapontine extension, enhancement, infiltrative margin, cystic/necrotic feature, hydrocephalus, and leptomeningeal seeding. Leptomeningeal dissemination was evaluated if whole-spine MRI data were available.
Radiotherapy characteristics (modality, dose, and target volume), chemotherapy regimen, and bevacizumab use for radiation necrosis were evaluated. Gross tumor volume (GTV) was defined as a T1-hypo (or iso) intense and T2-hyperintense lesion. Clinical target volume (CTV) was defined as GTV+0-2 cm margin, and planning target volume was defined as CTV+margin within 1 cm, according to the institutional policy.
3. Response evaluation
Treatment response was assessed using follow-up MRI acquired within 2 months after radiotherapy completion, and evaluation of clinical signs and symptoms. Treatment response was evaluated using T2-weighted images according to the Response Assessment in Pediatric Neuro-Oncology criteria and classified as follows [10]: complete response (CR; no evidence of disease), partial response (PR; ≥ 25% decrease in the two-dimensional [2D] products of the maximum perpendicular diameters), stable disease (SD; < 25% reduction or increase in 2D products of the maximum perpendicular diameters), and progressive disease (PD; ≥ 25% increase in the 2D products of the maximum perpendicular diameters or any new sites of disease). Clinical symptomatic responses were divided into three categories (improved, unchanged, and aggravated) based on the changes in clinical presentation recorded in the electronic medical records.
4. Statistical analysis
The date of diagnosis was based on the date of the first MRI scan revealing the evidence of DIPG, regardless of the pathologic diagnosis. OS was calculated from the date of diagnosis to the date of death or that of the last follow-up. Progression-free survival (PFS) was calculated from the date of diagnosis to that of the brain MRI, showing progression of disease, or to that of death. Independent sample t tests were used to compare continuous variables, such as baseline characteristics, between the two groups. Pearson’s chi-square test or Fisher exact test, as suitable, were used to compare categorical variables. The Kaplan-Meier method with log-rank test and Cox regression were used to analyze survival outcomes. Stepwise Cox proportional hazards regression was used for multivariable analysis of factors affecting OS (inclusion criteria, p < 0.05). All statistical tests were two-tailed. p-values of < 0.05 indicated statistical significance. The data were analyzed using IBM SPSS software ver. 20.0 (IBM Corp., Armonk, NY).
Results
1. Patient and treatment characteristics
Data from a total of 162 patients from 10 tertiary academic institutions were analyzed in this study. The median patient age was 7 years (interquartile range [IQR], 5.0 to 12.3 years) and only three patients were younger than 3 years. Symptoms related to cranial nerve palsy and cerebellar signs were observed in 63.6% and 66.7% of the patients, respectively. In contrast, pyramidal signs were relatively rare (32.1%). The median duration of symptoms was 4 weeks (IQR, 2 to 8). Histological diagnosis was established in 44 patients through biopsy or surgical resection. Most patients (90.9%) had a confirmed diagnosis of grade 3 glioma or higher. Eighteen patients were diagnosed with diffuse midline glioma, H3K27M-mutant, which corresponded to 85.7% of the patients who were histologically diagnosed after 2016. Information on molecular biomarkers was available for a very limited number of patients. MGMT data were available for 17 patients, including one with MGMT methylation. In addition, IDH1 mutation and 1p19q co-deletion were observed in three of 28 patients and 1 of 15 patients, respectively, for whom the data were available (Table 1).
Treatment details are summarized in Table 2. Intensity-modulated radiotherapy was used in 69 patients (42.6%), followed by 3D conformal radiotherapy in 63 (38.9%), proton therapy in 22 (13.6%), and 2D radiotherapy in eight patients (4.9%). The median dose of radiotherapy was 54 Gy (range, 30 to 62.5 Gy; IQR, 54 to 54 Gy). CTV was defined as GTV+less than 1 cm margin for 67 (41.3%) and GTV+1-2 cm margin in 68 patients (42%). Planning target volume was defined as CTV+3 mm margin in most patients. Concurrent chemotherapy was administered in 84 patients (51.9%) using temozolomide (TMZ) (48.1%) and regimens including vincristine, etoposide, or BCNU (1,3-bis(2-chloroethyl)-1-nitrosourea) (3.8%). Adjuvant chemotherapy was performed in 70 patients (43.2%) using TMZ (59/70 patients) and thalidomide, vincristine, ACNU (1-(4-amino-2-methyl-5-pirimi-dinyl)-methyl-3-(2-chloroethyl)-3-nitrosourea hydrochloride, or nimustine hydrochloride), or BCNU (11 patients). In the group that experienced disease progression after radiotherapy, 49 patients (35.3%) underwent salvage treatment. Of these patients, 45 underwent chemotherapy alone, two underwent re-radiotherapy with chemotherapy, and two underwent surgery.
2. Clinical outcomes
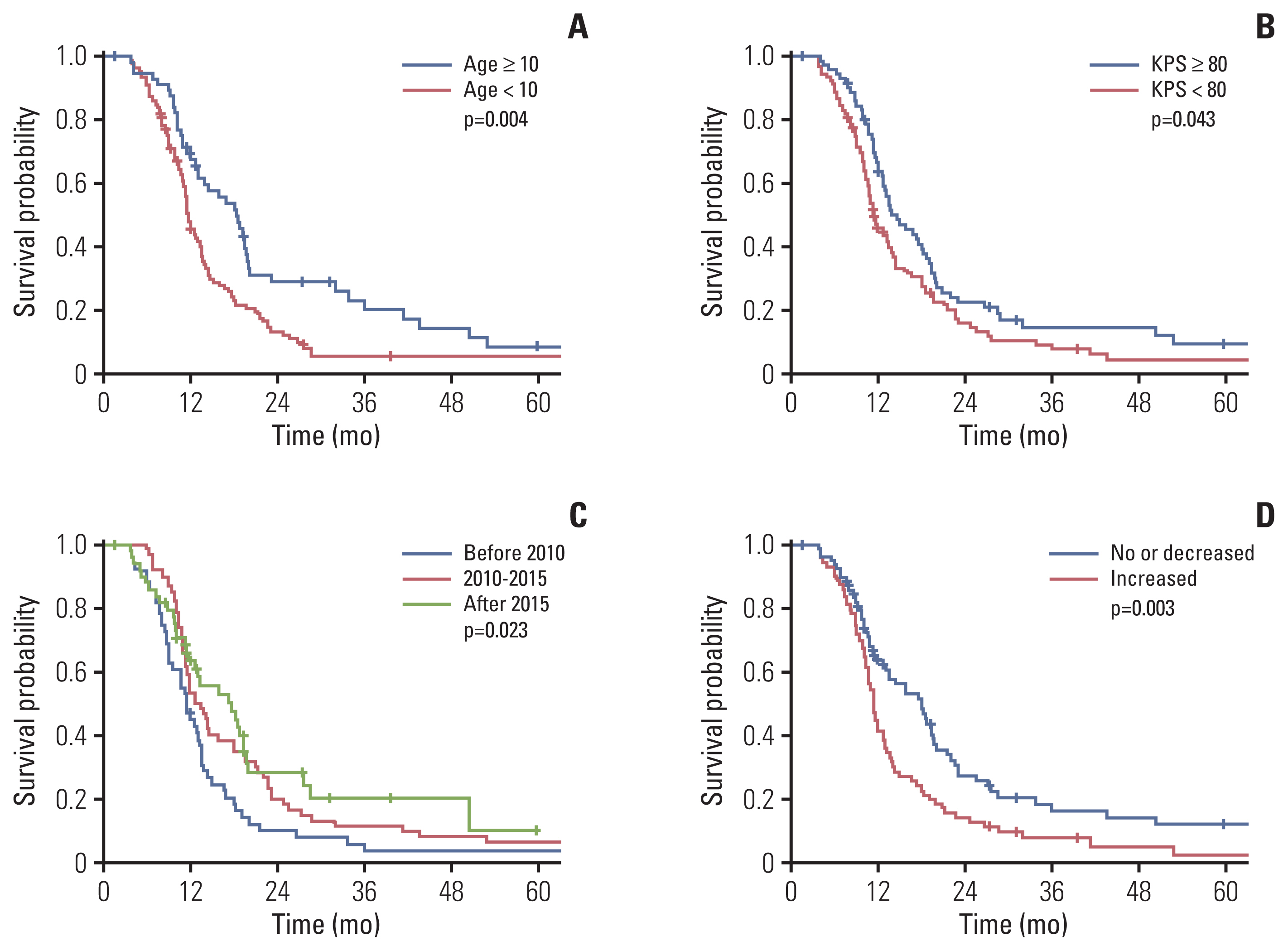
The median follow-up period was 10.8 months (IQR, 7.5 to 18.1). The 1-year and 2-year PFS rates were 26.3% and 8.4%, respectively, and the median PFS was 7.7 months (95% confidence interval [CI], 6.9 to 8.5). The 1-year and 2-year OS rates were 53.5% and 19.0%, respectively, and the median OS was 13.1 months (95% CI, 11.7 to 14.5 months). OS improved significantly over time (Fig. 1C). Median OS was 11.4, 13.5, and 17.6 months for patients treated before 2010, during 2010–2015, and after 2015, respectively, with statistically significant differences between the first and second groups (p=0.041), and between the first and third groups (p=0.011). The difference in OS between the second and third groups was not statistically significant (p=0.446). The 2-year OS rates of patients treated before and after 2010 were 10.2% and 23.2%, respectively (p=0.008).
Twenty-seven patients (16.7%) survived for ≥ 24 months, and their median OS was 43.6 months (95% CI, 24.3 to 62.9). Long-term survivors were more likely than short-term survivors to be older than 10 years (51.9% vs. 31.9%, p=0.047). Long-term survivors presented with higher rates of extrapontine extension (92.6% vs. 73.3%, p=0.035) and lower rates of cyst/necrosis (18.5% vs. 41.5%, p=0.025) than short-term survivors. In addition, more long-term than short-term survivors underwent salvage treatment (65% vs. 31.5%, p=0.005).
CR, PR, SD, and PD were observed in 4, 72, 49, and 35 patients, respectively. The overall response rate to radiotherapy was 47.5%. The median OS was not-reached, 10.7, 9.1, and 7.8 months for patients with CR, PR, SD, and PD, respectively (p=0.001). Symptomatic improvement was observed in 87 patients (53.7%); however, no change in symptoms was observed in 33 patients (20.4%), and worsening of symptoms was observed in 29 patients (17.9%). OS rates were 14.4, 9.8, and 8.0 months, respectively, in patients with improved, unchanged, and worsening symptoms (p=0.038). Infield intracranial failures were observed in 110 patients (67.9%), followed by outfield intracranial failures in 22 (13.6%) and extracranial seedings in four (2.5%) patients.
3. Prognostic factors
We performed univariate and multivariate analysis to investigate the prognostic factors related to OS, including clinical features, radiologic findings, and treatments (Table 3). Younger age (< 10 years), poor performance status at diagnosis, no extrapontine extension, cystic or necrotic feature, treatment before 2010, and post-radiotherapy necrosis were significantly associated with poor survival in univariate analysis (Table 3, Fig. 1). The 1-year OS was 44.3% for patients who had increased necrosis after radiotherapy and 63.9% for those who had stable or decreased necrosis (p=0.003) (Fig. 1D). In multivariate analysis, age < 10 years, poor performance status, treatment before 2010, and post-radiotherapy necrosis were independently associated with poor OS.
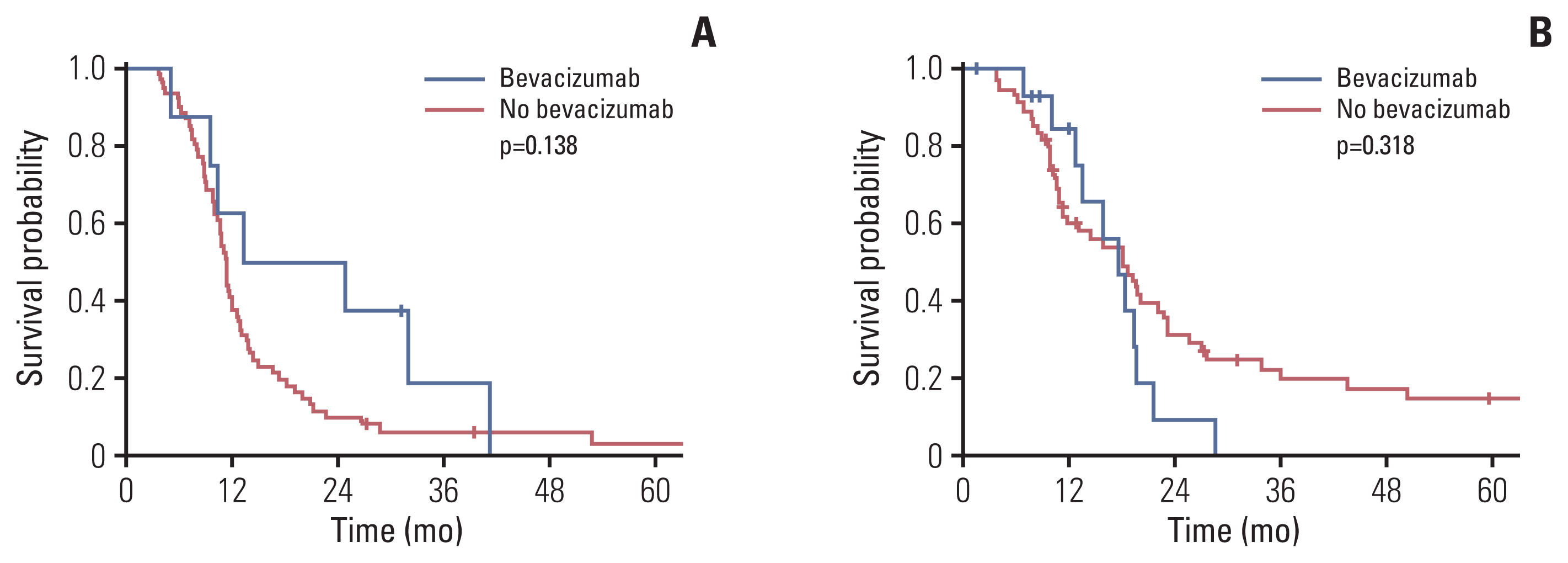
Bevacizumab was administered in 23 of 150 patients with available data. Patients who received bevacizumab showed a median survival of 18.3 months; the corresponding value for patients who did not receive this treatment was 12 months (p=0.386). Among patients who had increased post-radiotherapy necrosis, the median OS was 13.3 months and 11.4 months for those who did and did not use bevacizumab, respectively (p=0.138) (Fig. 2A). Among patients without or with decreased post-radiotherapy necrosis, the median OS was 17.6 months and 18.1 months for patients that did and did not use bevacizumab, respectively (p=0.318) (Fig. 2B). In addition, post-radiotherapy necrosis occurred more frequently, in patients younger than 10 years than in their counterparts (54.8% vs. 33.9%, p=0.013), in those with cystic or necrotic tumors on initial MRI (66.0% vs. 36.5%, p=0.001), and those who received adjuvant chemotherapy (57.6% vs. 38.6%, p=0.021) (S1 Table).
We investigated whether any specific radiation factors, including target volume, prescribed dose, and radiotherapy techniques, affected OS rates. After adjustments, radiotherapy techniques showed no effect on OS. We could not perform any meaningful analysis on dose impact, as the prescribed dose was 52–56 Gy in most patients (76.6%). The pattern of failure was comparable among CTV margin groups, since most failures represented infield progression (p=0.160) (Table 4).
Discussion
In this study, we investigated survival outcomes, and clinical, radiologic, and pathologic factors affecting prognosis in patients aged < 30 years treated with radiotherapy for DIPG in Korea. A previous study has shown that brainstem glioma is rare, and few affected patients present at any institution [11]. This multicenter study included 10 institutions and covered a period of approximately 20 years. In this study, the median survival time was 13 months, and the percentage of long-term survivors was 16.7%, which was comparable to previously reported results [3,8,12].
Consistent with previous findings, age ≥ 10 years and good performance status at diagnosis were prognostic factors in this study [1,3]. Furthermore, salvage treatment performed in these patients improved prognosis. Patients treated since 2010 had better outcomes than patients treated before 2010, even after adjusting for clinicopathological factors, imaging features, and treatment types. Patients treated at different times had comparable baseline characteristics, except for the radiotherapy techniques used (S2 Table). As the pattern of practice and dose of radiotherapy for DIPG remained unchanged throughout the study period, the improvement in survival may be accounted for by progress in service delivery, including improved socio-economic conditions and medical insurance reimbursement. Using Surveillance, Epidemiology and End Results data, Brandel et al. [13] reported that the survival of patients with grade 3 oligodendroglioma improved over time even after adjusting for treatment types. The authors also suggested that the improvement in survival may be due to changes in service delivery [13].
Historically, biopsy has not been recommended for diagnosis in this context, and surgical resection was hindered by the location and infiltrative nature of the tumor. This trend was reconfirmed through a recent survey from the European Society for Pediatric Oncology (SIOPE) brain tumor group [14]. Recent advances in brain stereotactic surgery have enabled tissue genomic analyses. The H3K27M mutation has been estimated to exist in 80% of DIPG cases [6,15], leading to a new disease entity, “diffuse midline glioma, H3K27M-mutant” being incorporated into the 2016 WHO classification of central nervous system tumors [5]. However, genetic or histologic findings are yet to translate to clinical practice [16]. In our study, biopsy and surgical resection were performed in 18.5% and 8.7% of patients, respectively. Moreover, data on molecular parameters were available in a very limited number of patients, precluding further analysis of molecular parameters. Therefore, previous DIPG prognostication studies were based on MRI findings, which can be easily obtained in the clinical environment [17–19].
We investigated MRI features associated with OS in this study. In univariate analysis, extrapontine extension of tumor, cystic/necrotic feature, and post-radiotherapy necrosis were associated with OS; among them, only post-radiotherapy necrosis was significantly associated with survival in multivariate analysis. A recent report of 357 patients included in an international DIPG registry showed that tumor extension beyond the pons, enhancement, and tumor necrosis were imaging features associated with OS in univariate analysis; however, none of these features was significant in multivariate analysis [18]. In addition, extrapontine extension was a poor prognostic factor for OS in that study, while it was associated with good prognosis in our study. Interpreting this result, we hypothesized that rapid tumor growth was associated with symptom development, which occurred before extrapontine extension development. In contrast, less aggressive tumors, associated with delayed symptom onset, were likely diagnosed once extrapontine extension was already present. Moreover, we observed that patients with extrapontine extension had fewer cystic or necrotic features than those without it (32.3% vs. 54.1%, p=0.016), which may suggest more indolent features of tumors with extrapontine extension. However, this finding was not statistically significant and further research is required to test this hypothesis.
Bevacizumab has shown efficacy in adult glioblastoma; however, little is known about its efficacy in pediatric patients with newly diagnosed or recurrent DIPG [20–22]. Hoffman et al. [1] reported that the use of bevacizumab at diagnosis improved the odds of long-term survival. Recently, Crotty et al. [22] reported a single-center study on the use of a maintenance regimen, which included TMZ, irinotecan, and bevacizumab following radiotherapy, showing prolonged survival in patients with DIPG, compared to single-agent TMZ. In our study, bevacizumab was used in 23 patients who presented with better OS outcomes than those treated with other regimens. When patients were divided according to the post-radiotherapy necrosis, we found that the difference in survival was more pronounced in patients with post-radiotherapy necrosis than in those without, although it was not significant. In light of these results, given the small number of patients in our study, we may suggest that the use of bevacizumab may be beneficial in pediatric patients with DIPG with increased post-radiotherapy necrosis. However, further well-designed trials are required to confirm these effects.
All patients included in this study underwent radiotherapy as their initial treatment; however, as the patients received a relatively homogenous dose of radiation (median, 54 Gy), we did not examine the impact of radiation dose on outcomes. Considering that most treatment failures were infield progression, and the pattern of failures was not affected by radiotherapy volume, we suggest that a margin of ≥ 1 cm to the target volume may not be beneficial. Similar results from 97 patients were reported by Tinkle et al. [23]; these authors concluded that no apparent survival or tumor-control benefit was achieved by extending the CTV margins beyond 1 cm.
Several studies have been conducted on neoadjuvant, concurrent, and adjuvant chemotherapy in addition to radiotherapy to improve the prognosis of patients with DIPG [24–26]. A phase II study evaluating the efficacy of chemoradiotherapy with TMZ followed by adjuvant TMZ conducted by the Children’s Oncology Group [9] reported 1-year event-free survival rate of 14%, which was lower than the corresponding rate of 21.9% observed in CCG-9941. Although similar findings on TMZ were reported in several prospective studies, herein, we found that concurrent or adjuvant TMZ remains part of practice at attending physicians’ discretion, although the evidence of its efficacy is scarce. These findings suggest that treatments that improve DIPG outcomes remain to be established.
This study had some limitations. First, due to the retrospective nature of this study, the present findings are likely subject to bias. However, given the low incidence of DIPG, prospective studies are not feasible. To minimize the effects of bias, we followed strict eligibility criteria to improve sample homogeneity. Second, the sample size was relatively small despite pooling patients treated at 10 tertiary institutions over a period of 20 years. However, treatment protocols and radiotherapy doses remained unchanged during the study period, reducing any impact of bias. Third, molecular data were scarce, precluding any meaningful analyses. This observation may reflect the clinical reality, where pathologic analyses are rarely performed outside of clinical trials.
In conclusion, radiotherapy protocols in the treatment for pediatric DIPG remained unchanged during the study period. The prognosis of DIPG patients remains poor despite slight improvement in survival over time. Radiation necrosis affects OS and should be reduced to improve outcomes. Furthermore, the use of bevacizumab for radiation necrosis may be helpful in some patients; however, further studies are required to examine this effect.