Introduction
The DNA damage response (DDR) pathway is an essential target for cancer treatment, and the effectiveness of DDR-targeted agents has been evaluated in a variety of cancers [1]. Biliary tract cancers (BTCs) are relatively rare cancers with few systemic treatment options and have poor prognosis [2]. Thus, novel treatment strategies are urgently needed to improve the outcomes of patients with advanced BTC. Up to 20% of patients with BTC exhibit alterations in DDR genes, including homologous recombination (HR) genes, which can be targeted by olaparib, a poly(ADP)-ribose polymerase (PARP) inhibitor [3,4]. Olaparib is an effective therapeutic strategy for patients with breast cancer, ovarian cancer, pancreatic cancer, and prostate cancer with germline BRCA mutations [5–8].
Olaparib disrupts single-strand break (SSB) repair by trapping PARP in DNA; this disruption of SSB repair at the replication fork provokes the conversion of unrepaired SSB to a more genotoxic single-ended double-strand break (seDSB) [9]. Repairing DNA double-strand breaks (DSBs) is essential for cell survival and is achieved via two mechanisms: non-homologous end joining (NHEJ) repair and HR repair [10]. The seDSBs can be faithfully repaired via the HR pathway in HR-proficient cancer cells, while HR repair-deficient cancer cells must rely on the less accurate NHEJ, resulting in increased genomic instability, which eventually leads to apoptosis [9,11]. This is why HR repair is well known as an olaparib resistance mechanism [12].
Furthermore, olaparib induces G2 arrest, which allows time to repair DNA damage [13]. G2 arrest with olaparib can be induced by a p53-dependent mechanism, wherein p53 induces p21, which prevents cyclin-dependent kinase 1 (CDK1) activation, thereby regulating G2 arrest [14]. CDK1 can also be inhibited in a p53-independent manner through the ATR-CHK1-CDC25 axis [15]. Although the p53-dependent pathway is well known to be involved in olaparib-induced G2 arrest, the p53-independent mechanism has not been sufficiently investigated.
The WEE1 checkpoint kinase has been shown to control both S-phase and G2/M progression by regulating CDK1/2 [16]. The CDK1 and cyclin B complex is the main factor that determines mitotic entry [17]. WEE1 blocks CDK1 activity and activates the G2/M cell cycle checkpoint to prevent unscheduled mitosis [16]. AZD1775, a WEE1 inhibitor, increases CDK1 activity by inhibiting WEE1, overriding the G2/M cell cycle checkpoint [18]. The premature cells in the G2 phase are forced to enter mitosis, resulting in mitotic catastrophe [19]. CDK1 also regulates HR repair through WEE1 inhibition, but the mechanism is not fully understood [20].
In this study, we aimed to investigate the combination of olaparib and AZD1775 as a new DDR-targeted therapy for BTC. We added AZD1775 to olaparib to regulate CDK1, the key molecule that causes G2 arrest in BTC, and focused on the effects of AZD1775 abrogating the G2 arrest and blocking the HR pathway to enhance the efficacy of olaparib.
Materials and Methods
1. Human cell lines and reagents
Ten human BTC cell lines were used in this study. SNU245, SNU308, SNU478, SNU869, SNU1079, and SNU1196 cells were purchased from the Korean Cell Line Bank (Seoul, Korea). HuCCT-1 and TFK-1 cells were obtained from the RIKEN BioResource Center (Ibaraki, Japan). SNU2670 and SNU2773 cell lines were established from patient-derived cancer cells, as previously described [21]. All cell lines were cultured in RPMI-1640 medium (Welgen Inc., Gyeongsan, Korea) containing 10% fetal bovine serum and 10 μg/mL gentamicin and were maintained at 37°C in a 5% CO2 atmosphere. WEE1 inhibitor (AZD1775) and PARP inhibitor (olaparib) were provided by AstraZeneca (Macclesfield, Cheshire, UK).
2. Cell viability assay
Cell viability was measured using MTT assay. First, cells were plated in 96-well plates and incubated overnight at 37°C before the addition of olaparib and AZD1775 alone or in combination. Five days later, 50 μL of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Sigma-Aldrich, St. Louis, MO) was added to each well and incubated for 4 hours at 37°C. The MTT solution and medium were removed, and 150 μL DMSO was added to each well. The absorbance was measured at 540 nm using a VersaMax Microplate Reader (Molecular Devices, San Jose, CA), and the cell viability was calculated.
3. Western blotting
Cells were collected after treatment with olaparib, AZD1775, or both for 24 hours and 72 hours. The cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in RIPA buffer containing protease inhibitors for 30 minutes on ice. Proteins were obtained by centrifugation at 13,000 rpm for 20 minutes, and equal amounts were used for western blotting. Primary antibodies against the following proteins were purchased from Cell Signaling Technology (Beverley, MA): PARP (#9532), caspase-3 (#9662), caspase-7 (#9492), WEE1 (#4936), p-cdc2-Tyr15 (#9111), CtIP (#9201), NBS1 (#3002), p-ATM-Ser1981 (#5883), p-Chk2-Thr68 (#2661), ATR (#2790), p-CHK1-Ser345 (#2341), p-CDC25C-Ser216 (#9528), p53 (#9282), p-p53-Ser15 (#9284), p21 (#2947), and cyclin B1 (#4138). GAPDH (#sc-25778) and RAD51 (#sc-398587) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The p-CDK2-Tyr15(#ab-76146) antibody was purchased from Abcam (Cambridge, UK). Anti–γ-H2AX antibody (#05-636) was obtained from Millipore (Billerica, MA). p-ATR-Thr1989 (#GTX-128145) was purchased from GeneTex (Irvine, CA), and secondary antibodies were purchased from Thermo Fisher Scientific (Waltham, MA).
4. Cell cycle analysis
Treated cells were trypsinized, harvested, and fixed with 70% ethanol at 20°C for more than 2 days. Ethanol was removed, and the cells were resuspended in fluorescent activated cell sorting (FACS) buffer and incubated with 7 μL of RNase A (Invitrogen) (20 mg/mL) for 10 minutes at 37°C. Samples were stained with 13 μL of propidium iodide (PI; Sigma-Aldrich) and analyzed on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
5. Colony-formation assay
Cells were seeded on a 6-well plate and treated with olaparib, AZD1775, or both. Cells were cultured at 37°C in a 5% CO2 incubator for 10 days. The colonies were rinsed with PBS and stained with Coomassie blue for 3 hours. The stained colonies were counted using the CellCounter plugins.
6. Annexin V/PI apoptosis assay
Cells were seeded in 60-mm dishes and treated with olaparib, AZD1775, or both. Treated cells were harvested with trypsin and resuspended in binding buffer (#556547, BD Biosciences). The apoptosis assay was performed by double staining with 1 μL Annexin V-FITC and 2 μL PI (#556547, BD Biosciences). and incubated for 15 minutes at RT in the dark. The cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences). Cells in early and late apoptosis were defined as Annexin V-FITC-positive/PI-negative and Annexin V-FITC-positive/PI-positive cells.
7. Phospho-histone H3 staining assay
Treated cells were harvested with trypsin and fixed with 70% ethanol for at least 4 hours at 20°C. After washing with staining buffer (#420201, BioLegend, San Diego, CA), 20 μL of p-HH3 antibody (#558217, BD Bioscience), and 80 μL of staining buffer were added to each sample for 20 minutes at room temperature. Cells were washed with staining buffer and incubated with 200 μL of staining buffer and 2 μL of RNase A for 10 minutes at 37°C. Next, 5 μL of PI was added, and the cells were analyzed using a FACSCalibur flow cytometer. Each experiment was repeated three times.
8. Immunofluorescence assay
For RAD51 and γ-H2AX foci analysis, cells were seeded on a confocal dish, incubated for 48 hours, and then treated for 24 hours. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. Cells were incubated with a rabbit monoclonal RAD51 (#sc-398587) antibody and mouse monoclonal γ-H2AX (#05-636) antibodies overnight at 4°C. Samples were stained with fluorochrome-conjugated secondary antibody, Alexa Fluor 488 anti-rabbit (#2147635) and Alexa Fluor 594 anti-mouse (#2179228) (Invitrogen, Carlsbad, CA), and counterstained with DAPI (Sigma-Aldrich). Immunofluorescence was visualized using a Zeiss LSM 800 laser scanning microscope at 40× magnification.
9. Alkaline comet assay
Treated cells were harvested, resuspended at 5×105 cells/mL in ice-cold PBS, and combined with molten LMAgarose at a ratio of 1:10. Next, the samples were moved onto comet slides and incubated at 4°C in the dark for 40 minutes. After dipping the slides in pre-cooled lysis solution at 4°C for 40 minutes, the slides were gently moved in freshly prepared alkaline unwinding solution (200 mM NaOH, 1 mM EDTA, pH > 13) for 30 minutes at room temperature in the dark. Electrophoresis was conducted for 30 minutes, and the samples were dried at room temperature overnight. Next, 100 μL of diluted SYBR Green staining solution was dropped onto each circle of agarose, and the samples were covered with a coverslip. The tail moment and intensity were measured using the Comet Assay IV program (Andor Technology, Belfast, UK). Three independent experiments were performed for each condition.
10. siRNA transfection
After seeding cells in 100-mm dishes and incubated at 37°C with normal medium. SiRNA specific for WEE1 gene and negative control were purchase form Genolution (Seoul, Korea). Cells were transfected with siWEE1 at 50 nM final concentration for 6 hours with serum free medium. After 6 hours, it was changed to normal medium and incubated for 24 hours. We used the sequence of specific siRNAs as follows: negative control, 5′-CCUCGUGCCGUUCCAUCAGGUAGUU-3′; siWEE1_#1, 5′-GCAUUCUCAUGUAGUUCGAUU-3′, and siWEE1_#4, 5′-CUAUGUCAGGCUUUCAU-CUUU 3′.
11. In vivo experiments
Four-week-old female athymic nude mice were purchased from Orient Bio Inc. (Seongnam, Korea). SNU869 cells (1×107 cells) were subcutaneously injected into each mouse. When the tumor volume reached 250 mm3, the mice were randomly divided into four groups of five mice each. Olaparib (50 mg/kg) and AZD1775 (25 mg/kg) were administered orally once a day for 4 weeks (5 days on/2 days off), and the control group was treated with vehicle (0.5% methyl cellulose, Sigma-Aldrich) via oral gavage. Body weight and tumor size were measured every other day. The tumor volume was calculated using the following formula: tumor volume=[(width)2×height]/2.
12. Immunohistochemistry
The isolated xenograft tumor tissues were fixed in 4% paraformaldehyde and embedded in paraffin. The paraffin blocks were sectioned on glass slides, and the slides were deparaffinized and dehydrated. The samples were detected by immunohistochemical staining with p-CDK1-Y15 (AP0016) antibody, and proliferation was evaluated using an anti-Ki67 antibody (GeneTex Inc.). Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay-based ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore) was used to evaluate apoptosis.
13. Statistical analysis
Statistical analyses were conducted using SigmaPlot ver. 10.0 (Systat Software Inc., San Jose, CA). Experimental data are presented as the mean±standard error. All statistical tests were two-sided. Differences were considered statistically significant at p < 0.05. The half-maximal inhibitory concentration (IC50) of the agents was also determined using the SigmaPlot software. The effects of combined drug were analyzed by calculating the combination index (CI) using CalcuSyn software (Biosoft, Cambridge, UK). CIs of < 1, 1, and > 1 to indicate synergistic, additive, and antagonistic effects, respectively.
Results
1. Olaparib-induced DNA damage and simultaneously activated G2/M checkpoint
To assess the anti-proliferative effects of olaparib in BTC cell lines, the MTT assay was performed after treatment with olaparib for 120 hours. The anti-proliferative effect of olaparib in 10 BTC cells was modest. The IC50 in four cell lines (SNU869, HuCCT-1, SNU2773, and SNU478) was between 2.96–6.49 μM, while that of the other six was higher than 10 μM (Fig. 1A).
Two cell lines were selected for further experiments based on the IC50 of olaparib in the MTT assay. SNU869 showed the lowest IC50, and SNU308 showed an IC50 higher than 10 μM. We observed that olaparib-induced DNA damage and apoptosis by western blotting. The expression of γ-H2AX, cleavage of PARP, cleavage of caspase-7, and cleavage of caspase-3 were induced in both cell lines (Fig. 1B). Furthermore, we performed cell cycle analysis and observed that the sub-G1 phase increased in SNU869 and SNU308 by the olaparib. SNU869 was sensitive to olaparib than SNU308, the sub-G1 phase increased potently (Fig. 1C).
We also observed an increase in G2/M phases in two BTC cell lines in a dose-dependent manner (Fig. 1C). Olaparib increased the expression or phosphorylation of multiple proteins involved in G2/M checkpoints, including WEE1, p-CDK1, and cyclin B1. The protein expression of p-ATR, p-ATM, p-CHK1, p-CHK2, and p21 were also increased in the two cell lines, and p-p53 was increased in SNU308 cells. These data suggest that olaparib activates p-CDK1 and causes G2 arrest in two cell lines through both p53-dependent and p53-independent pathways (Fig. 1D). Furthermore, the expression of CtIP, RAD51, and NBS1 was increased in cells treated with olaparib, implying that DNA DSB was repaired by the HR pathway (Fig. 1D).
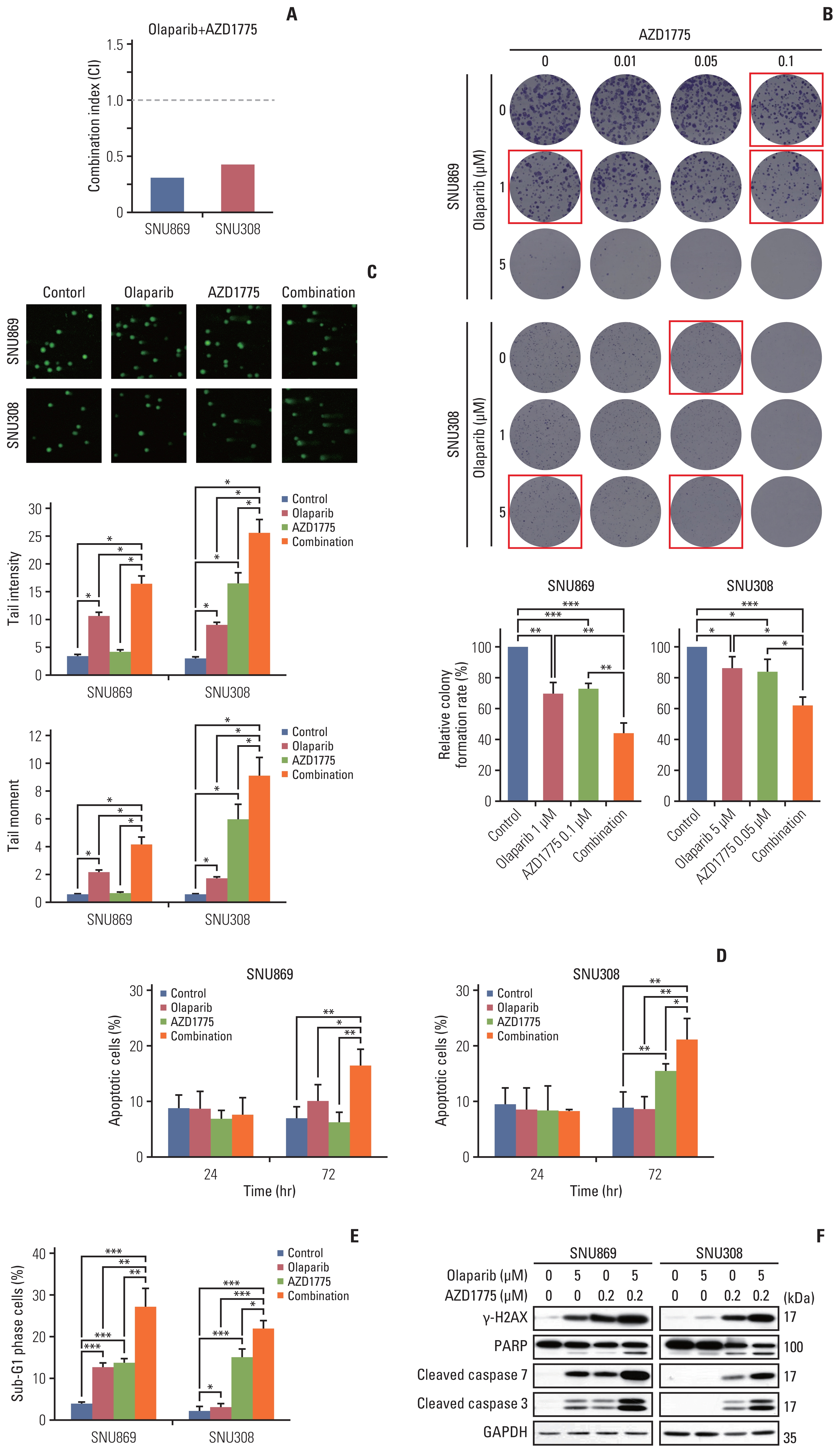
2. Combination therapy with olaparib and AZD1775 induced more potent DNA damage and apoptosis
We observed that olaparib-induced G2 arrest, allowing cancer cells the time to repair DNA damage (Fig. 1C and D). For this reason, we combined olaparib with AZD1775 to inhibit CDK1. To assess the anti-proliferative effects of olaparib and AZD1775, we performed the MTT assay, in which the CI values were lower than 0.5, and synergy was found in two cell lines (Fig. 2A). The colony-forming assay also showed an increase in the anti-proliferation effect in the combination treatment with different doses of olaparib and AZD1775 (Fig. 2B). We performed the comet assay to analyze the increase in DNA damage after treatment with olaparib, AZD1775, and their combination for 72 hours. The tail intensity and tail moment increased significantly after combination treatment in the SNU869 and SNU308 cell lines (Fig. 2C).
In the annexin V and cell cycle analyses, the annexin V-positive cells and the sub-G1 proportions were higher in the combination therapy than in either monotherapy (Fig. 2D and E). We observed the increase of apoptotic cells at 72 hour-combination therapy, not at 24-hour treatment (Fig. 2D). These observations indicate that the apoptosis rate was higher in the combination therapy group than in the olaparib or AZD1775 monotherapy group. In agreement with this result, the expression of γ-H2AX, cleavage of PARP, cleavage of caspase-7, and cleavage of caspase-3 were all increased in the combination therapy group than in the monotherapy groups (Fig. 2F). These data showed that adding AZD1775 to olaparib resulted in increased DNA damage and apoptosis in BTC cells.
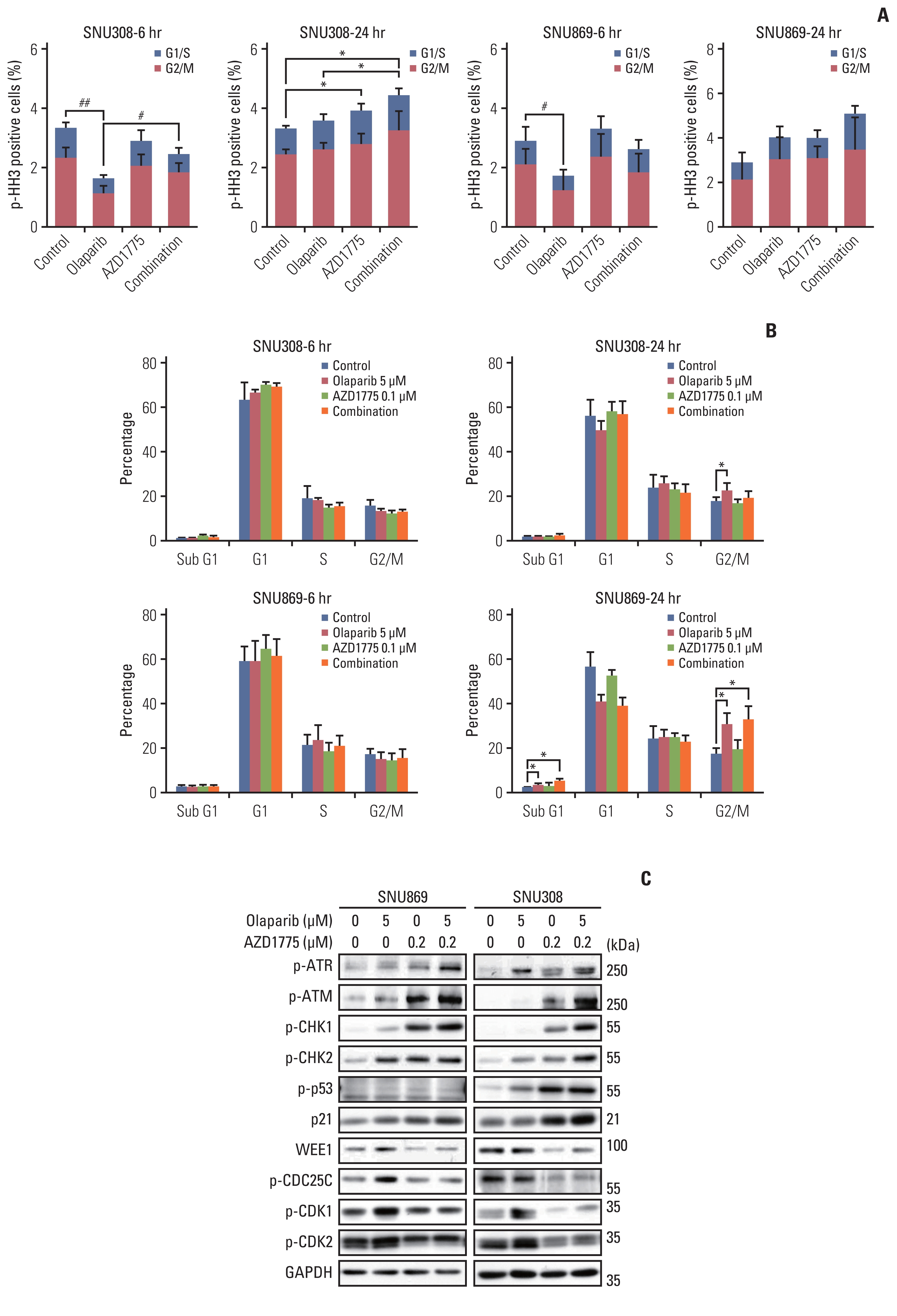
3. AZD1775 abrogated G2 arrest and induced early mitotic entry
As olaparib-treated cells induced G2 arrest, we hypothesized that AZD1775 might increase the sensitivity of olaparib by inducing early mitotic entry of cancer cells. We therefore performed a phospho-histone H3 staining assay to detect the increase in mitotic cells for 6 hours and 24 hours. The proportion of p-HH3-positive cells decreased at 6 hours after treated with olaparib in G2/M phase, but, when co-treated with AZD1775, cells undergo mitosis (Fig. 3A). In SNU308, the proportion of p-HH3-positive cells increased by AZD1775 alone or in combination at 24 hours, indicating an increased percentage of cells in the mitotic phase (Fig. 3A). Additionally, cell cycle showed that olaparib increased the G2/M phase in 24 hours, although the mitotic cells did not increase in two cell lines. In combination therapy, G2/M phase was not increased more than olaparib, but the mitotic cells were more increased in SNU308 (Fig. 3B). Taken together, olaparib-induced G2 arrest, and AZD1775 abrogated G2 arrest and induced mitosis. SNU869 was also statistically increased mitotic phase by higher AZD1775 (0.2 μM) (data not shown).
Combination treatment with olaparib and AZD1775 increased p-ATR, p-CHK1, p-p53, and p21, but WEE1, p-CDK1, p-CDK2, and p-CDC25C expression decreased due to AZD1775 (Fig. 3C).
These data showed that the inhibition of WEE1 overrode olaparib-induced G2 arrest and forced olaparib-treated cells to enter mitosis.
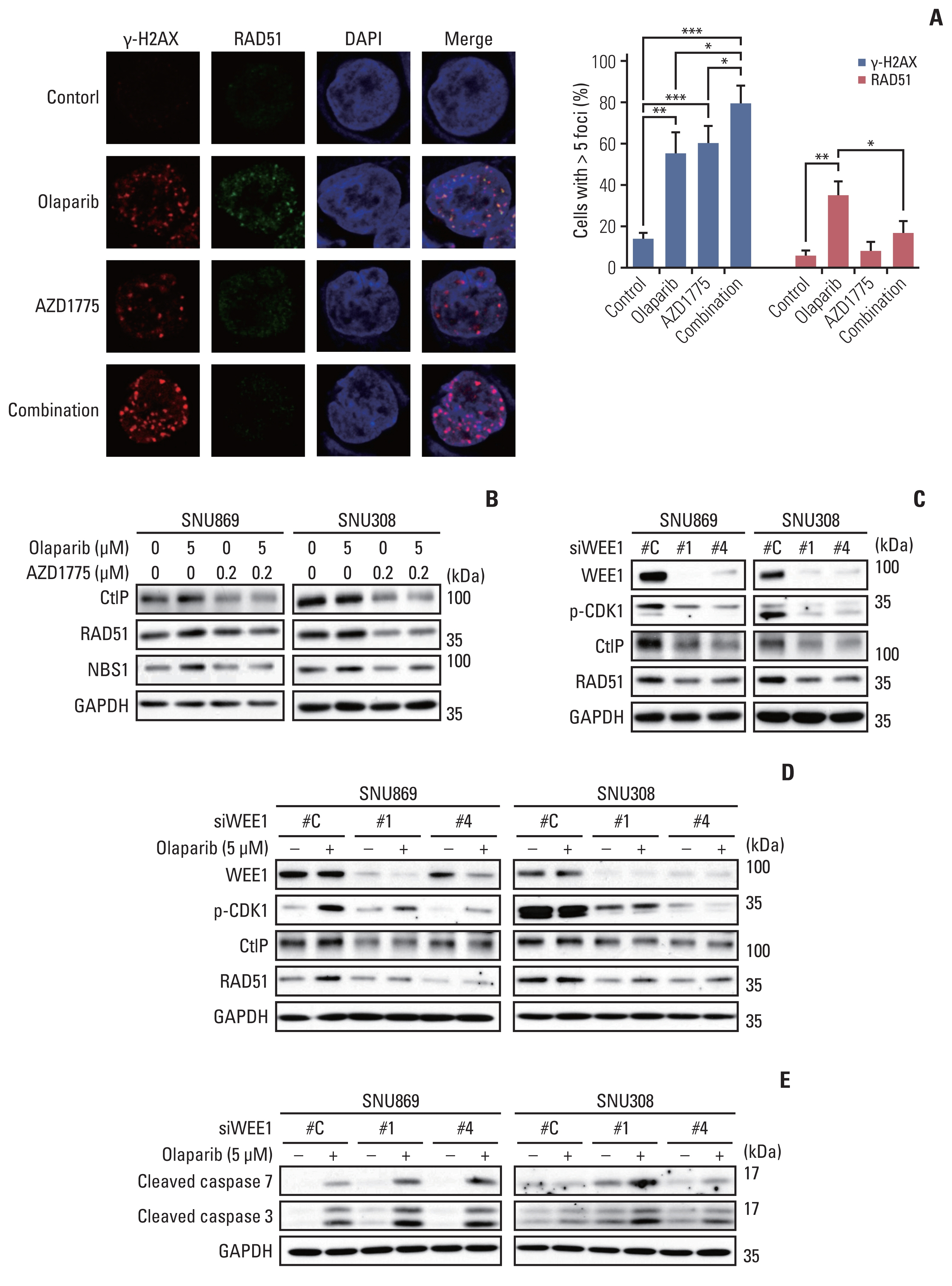
4. AZD1775 disrupted HR repair and enhanced the effects of olaparib
An immunofluorescence assay was performed to investigate the HR-disrupting effects of AZD1775. The number of γ-H2AX foci formation was more significantly increased by combination treatment with olaparib and AZD1775 than by monotherapy with either drug. On the other hand, olaparib-induced RAD51 foci formation was decreased by the combination treatment with AZD1775 (Fig. 4A). Consistent with this finding, western blotting data revealed decreased expression of RAD51 in the AZD1775 group. We also showed that AZD1775 downregulated CtIP and NBS1 level, keeping this condition in combination treatment (Fig. 4B). Although the AZD1775 mono-treated cells slightly increased RAD51 accumulation because of DNA damage, AZD1775 decreased RAD51 protein level and disrupted HR repair. Our findings suggest that combination treatment with AZD1775 induced more DNA damage than olaparib monotherapy, while AZD1775 diminished DNA damage repair by blocking the HR repair process.
Additionally, we were transfected the cells with siWEE1 to test whether WEE1 inhibition could also have the same effect of AZD1775. Transfection with siWEE1 showed effective knockdown of WEE1 expression, and reduced the expression of p-CDK1, RAD51, and CtIP proteins (Fig. 4C). After transfected with siWEE1, we treated olaparib for 24 hours. The expression of p-CDK, RAD51, and CtIP proteins were not increased more than olaparib-treated control cells (Fig. 4D). Furthermore, we treated olaparib for 72 hours after siWEE1 transfection and examined the increase of apoptosis markers expression such as cleavage of caspase 3 and cleavage of caspase 7 (Fig. 4E). These results indicated that WEE1 inhibition decreased p-CDK1 and HR repair protein expression and induced synergistic apoptotic effect with olaparib.
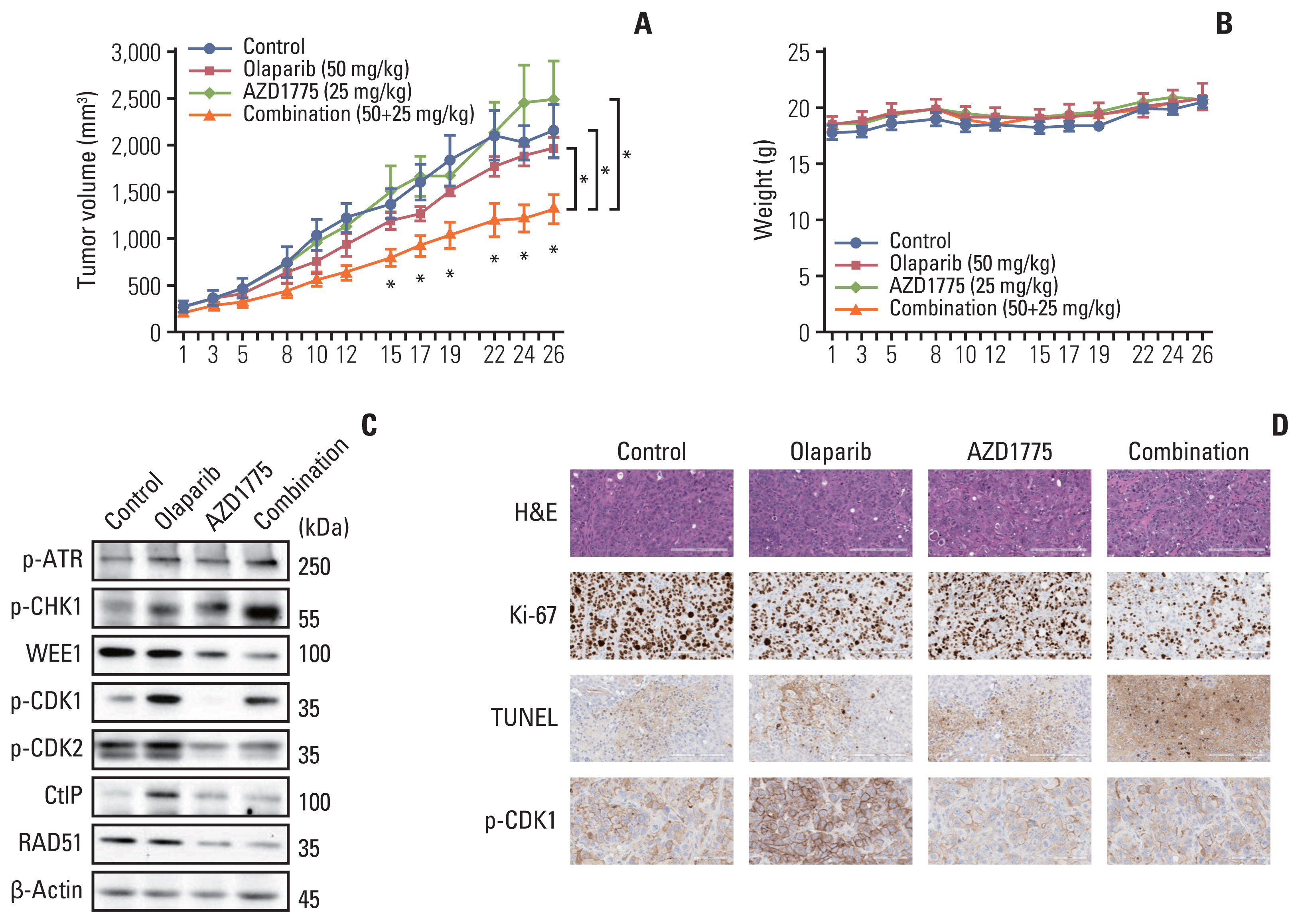
5. Combination treatment of olaparib with AZD1775 showed anti-tumor growth effects in a xenograft mouse model
To determine whether olaparib plus AZD1775 could exert an anti-tumor effect in vivo, we established a SNU869 xenograft model. In in vivo test, we could evaluate the safety, toxicity, and efficacy of drugs. Combination therapy significantly reduced tumor growth compared with that in the olaparib monotherapy and control groups (Fig. 5A). In vivo test was performed 4 weeks, which is much longer than in vitro test. We showed reliable drugs combination effect compared with in vitro test. The combination treatment was also well-tolerated (Fig. 5B). Similar to the in vitro results, p-ATR and p-CHK1 levels were increased by combination therapy, whereas WEE1, p-CDK1, p-CDK2, CtIP, and RAD51 were decreased by the addition of AZD1775, compared with those in the olaparib monotherapy group (Fig. 5C). Immunohistochemical staining revealed that the proliferation marker ki-67 was decreased in tumor tissues by the combination of olaparib and AZD1775. Furthermore, apoptosis was increased in combination therapy, which is represented by an increase in TUNEL staining. When we quantified the level of p-CDK1, it was decreased in combination therapy, which is consistent with our in vitro data (Fig. 5D). These in vivo results supported our in vitro data that dual blocking of PARP and WEE1 improved anti-tumor effects compared with those of monotherapy.
Discussion
In this study, we’ve found that olaparib combined with AZD1775 can induce synergistic anti-tumor effects against BTC. Olaparib monotherapy had a modest anti-proliferative effect in 10 BTC cell lines. After olaparib treatment, DNA damage and apoptosis were increased; however, G2 arrest was simultaneously induced, enabling for DNA damage repair. Previous studies have shown that olaparib induces G2 arrest through the p53-p21-CDK1 axis [14]. In our previous report, the basal level of p53 was relatively low in SNU869 cells and high in SNU308 compared with that in other BTC cell lines [22]. In this study, although the p53 and p-p53 levels were different between the two BTC cell lines (SNU869, SNU308), we observed that the phosphorylation of CDK1 was increased and G2 arrest was induced in olaparib-treated cells.
It is known that in response to DNA damage, the G2 checkpoint is regulated by two main mechanisms: p53-p21-CDK1 or CHK1/2-CDC25C-CDK1 [23]. The CDC25C is generally known to compete with WEE1 kinase to activate CDK1/cyclin B1 complex in a bistable system. After DNA damage, the CDC25C could be phosphorylated by multiple signaling pathways, and the WEE1 also could phosphorylate CDC25C [24]. Because the CDK1/cyclin B1 complex is a key molecule regulating G2/M transition, AZD1775 treated-cancer cells abrogated the G2/M checkpoint, leading to early mitotic entry. Furthermore, early mitosis can cause mitotic catastrophe and subsequently lead to apoptosis [16,18]. Based on findings of G2 arrest with olaparib monotherapy, we combined olaparib with a WEE1 inhibitor to disrupt DNA repair at the G2 phase by inhibiting CDK1 phosphorylation. Consistent with other reports, in our BTC study, we showed that AZD1775 abrogated G2 arrest by decreasing p-CDK1 expression and increasing p-HH3 positive cells.
Olaparib induces DNA DSB; however, damaged DNA was mostly repaired by the HR pathway [12]. We showed that olaparib-induced modest anti-proliferative effect, which presumably indicate the engagement of functional HR repair. Repair of double-stranded DNA damage through the HR is also known as one of the resistance mechanisms of olaparib [12]. In addition, previous studies have reported that the expression of RAD51 and CtIP regulates the sensitivity of breast cancer cells to olaparib [25,26]. In our study, we observed that DSB repairs through the HR pathway, which was indicated by an increase in HR repair protein expression. We also observed that AZD1775 disrupted the HR pathway by decreasing the recruitment of RAD51 on DNA damage sites. RAD51 protects single-stranded DNA and repairs DNA damage. A recent study showed that CDK1 regulates the phosphorylation of RAD51 during HR repair [27]. We showed that AZD1775 decreased RAD51 expression.
Additionally, CtIP initiates HR repair by interacting with the MRE11-RAD50-NBS1 complex and by promoting DNA end resection [28,29]. CDK2 regulates CtIP, and plays an important role in the interaction with the MRE11-RAD50-NBS1 complex to initiate HR [28,29]. To the best of our knowledge, our study is the first to show that AZD1775 regulates CtIP through CDK2. The combination of olaparib and AZD1775 decreased DSB repair and eventually increased cell death. Taken together, the AZD1775 regulates HR repair as well as cell cycle checkpoint, so it might be a good rationale for the enhanced combination effect with olaparib.
Even though BTCs harbor DDR alterations in significant amount of cases, DDR-targeted strategies for new drug development have not been much explored so far in BTC. We’ve pre-clinically tested the ATR inhibitor monotherapy, ATR inhibitor in combination with cisplatin, ATR inhibitor in combination with WEE1 inhibitor, PARP inhibitor in combination with ATR inhibitor, etc. in BTC [22,30]. For clinical development of DDR-targeted strategies in BTC, we are now conducting clinical trials using DDR-acting agents in advanced BTC patients (NCT04298008, NCT04298021). Further studies are needed for proving the concept of DDR-targeting strategies in BTC.
This study is the first to give preclinical evidence that dual-inhibition of PARP and WEE1 has a synergistic anti-tumor effect in BTC. This combination strategy of PARP inhibitor with WEE1 inhibitor could have the potential to be further developed clinically in patients with advanced BTC.