Introduction
Gastric cancer (GC) ranks as the third leading cause of cancer-related death worldwide. Every year, approximately one million people are diagnosed with GC, over half of which reside in Asia [1]. Surgical resection remains the most effective treatment for early and some advanced forms of GC, and over the last decade, chemotherapy has been considered a standard therapeutic regimen for patients with advanced or metastatic GC [2]. However, most patients with GC will eventually metastasize or relapse and the prognosis is still unsatisfactory [3]. Therefore, there is an urgent need to explore novel prognostic biomarkers to increase the accuracy of prognosis prediction and seize therapeutic opportunities for GC patients.
GC has substantial biological differences between Asian and non-Asian populations [4], which makes it difficult to have a unified predictive measure for all people. Due to the complex biological and molecular mechanisms underlying GC, traditional predict methods relying on clinical data such as serum examination, imaging examination, and pathologic information are limited.
In this genomic era, high-throughput platforms such as sequencing and microarrays play an increasingly important role in the field of oncology and make precision medicine possible. Using the hepatocellular carcinoma data set of The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), Long et al. [5] established a prognostic model for overall survival (OS) prediction, suggesting that these approaches and data have a wide range of clinical applications. Hou et al. [6] analyzed the GC expression profile data in the GEO database through Lasso regression analysis, and obtained 11 genes related to prognosis, and thought that the selection of more than five genes can predict the prognosis. Cheong et al. [7] established a 4-gene chemotherapy prediction model based on the expression profile data of GC after D2 surgery, which can assess whether patients can benefit from postoperative adjuvant chemotherapy. The incidence of GC in Asian populations is much higher than that of other races. However, at present, there is no prognostic model based on comprehensive clinical data and gene expression data, which can make a more accurate judgment on the prognosis of Asian GC patients.
In our study, we used a GEO dataset from Korea to establish a four-gene prognostic model, including mRNA processing factor 2 (RBPMS2), regucalcin (RGN), pleckstrin homology domain containing S1 (PLEKHS1), and cancer/testis antigen 83 (CT83). All patients were classified into a low-risk group and a high-risk group. The prognostic model was validated in the Asian GC cohort in the TCGA database. Finally, a nomogram including clinical characteristics and prognostic model was established for OS prediction. As a whole, these predictive methods will help stratify Asian patients with GC more accurately and promote more precise treatment for all of them.
Materials and Methods
1. Microarray data
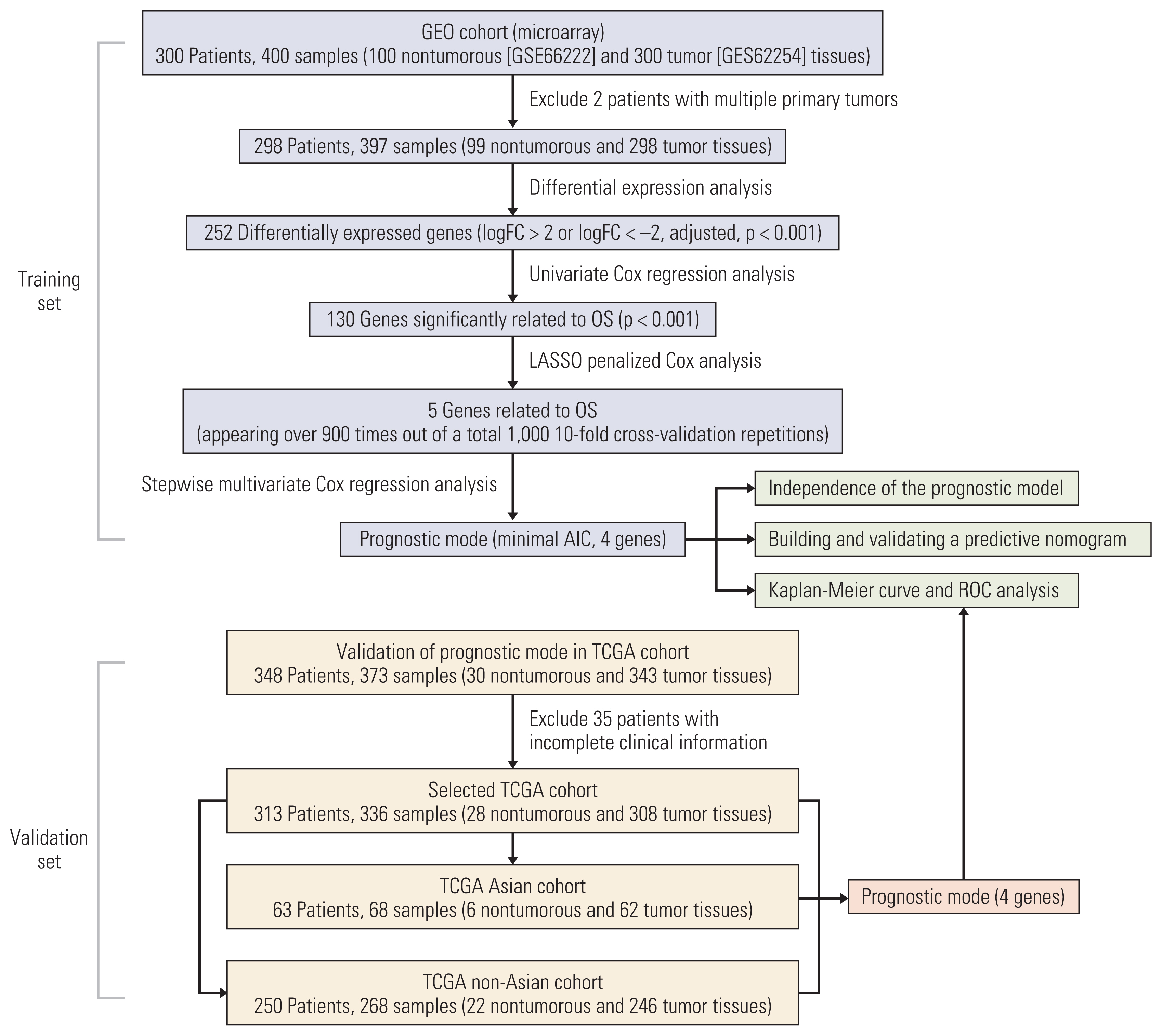
The gene expression profile matrix of GSE66229 was downloaded from the GEO database, and the clinical information was obtained from the corresponding published literature [8]. GSE66229 is composed of the GSE62254 and GSE66222 datasets, which were based on the GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array, Santa Clara, CA). The GSE62254 dataset contains 300 GC samples, and the GSE66222 dataset contains 100 adjacent nontumorous gastric tissues. Patients with GSE66222 are all included in GSE62254. We excluded two patients with multiple primary tumors. A total of 298 patients with 397 samples (99 adjacent nontumorous and 298 tumor tissues) were selected for further analyses.
2. RNA-sequencing data
The RNA-sequencing data and the corresponding clinical information used for validation were downloaded from the TCGA database. Among these GC patients, there were 348 patients with 373 samples (30 adjacent nontumorous and 343 tumor tissues). We excluded 35 patients with incomplete clinical information. Finally, 313 patients with 336 samples (28 adjacent nontumorous and 308 tumor tissues) were selected as the TCGA global cohort. According to the patients’ race, the cohort was further divided into an Asian cohort and a non-Asian cohort. The Asian cohort contained 63 patients with 68 samples (6 nontumorous and 62 tumor tissues), and the non-Asian cohort contained 250 patients with 268 samples (22 nontumorous and 246 tumor tissues).
3. Identification of differentially expressed mRNA
Firstly, we obtained the original mRNA expression profiles of GC from the GSE66229 dataset and merged them into one file. The RMA algorithm is used to normalize and Log2 transform the expression data in R environment. The average value of gene expression was taken when duplicate data were found. Genes with an average expression value > 1 were retained, while the low abundance sequencing data were deleted. The mRNA microarray includes 20,486 mRNA expression profiles. Next, we calculated the identification of differentially expressed genes (DEGs) with the help of the Limma package [9], where genes with logFC > 2 or logFC < −2 and adjusted p < 0.001 were considered for subsequent analysis, the cutoff value of LogFC was also used in other study [10]. The Pheatmap package and Volcanomap package were applied to describe the DEGs via the R language.
4. Establishment and verification of the prognostic model
Univariate, Lasso-penalized, and multivariate Cox regression analyses were performed to reduce candidate genes and explore the correlation between the mRNA expression levels and patient OS.
In the univariate Cox regression, mRNA expression was considered to be significant when the p < 0.001. Then, we conducted Lasso-penalized Cox regression to further reduce the number of genes. For the selection operator of Lasso-penalized Cox regression, we sub-sampled the dataset that was replaced one thousand times and selected markers with a repetition frequency greater than 900. The tuning parameters were determined by the expected generalization error, which was estimated by 10-fold cross-validation and information-based Akaike Information Criteria/Bayesian Information Criteria. To ensure that the error was within one standard error from the minimal (MSE), the maximum value of the lambda was adopted, which was defined as “1-MSE” lambda. Then, the expression data of candidate genes was standardized through the scale method in R Software (R Foundation for Statistical Computing, Vienna, Austria). To evaluate individual genes as independent prognostic factors for OS, we performed a multivariate Cox regression analysis. The stepwise approach was applied to further select the best model. Ultimately, a prognostic model based on four genes was established. The risk score was based on a linear combination of the expression levels of individual genes multiplied by their multivariate Cox regression coefficients (β): risk score=(expression level of RBPMS2×β)+(expression level of RGN×β)+(expression level of PLEKHS1×β)+(expression level of CT83×β). The X-tile software was used to determine the optimal cutoff value [11]. Kaplan-Meier survival analyses were performed for the low-risk group and the high-risk group and the log-rank test was used to compare survival rates. A time-dependent receiver operating characteristic (ROC) curve was performed to assess the predictive power of the predictive model. Independence analysis of diagnostic models with other clinical characteristics was conducted by univariate and multivariate Cox regression analysis.
The relationship between the expression of the four genes and OS in the GSE62254 dataset was verified in a Kaplan-Meier plotter (KmPlot, https://kmplot.com/analysis/index.php?p=service&cancer=gastric). Validation of the prognostic model was performed using the TCGA database. The expression data of the four genes was standardized by the scale method as well in validation cohorts.
5. Validation of the expression levels of the four genes in TCGA cohorts
We extracted the expression levels of the four genes in the GEO cohort, TCGA global cohort, and TCGA Asian cohort to further explored the expression patterns of the four genes. The different expression patterns between the low-risk group and the high-risk group in the three cohorts were analyzed by Wilcoxon signed-rank test. And the analysis was performed with statistical software GraphPad Prism ver. 7.0 (GraphPad Software, Inc., San Diego, CA). The p-values are two-sided, and p < 0.05 was considered statistically significant.
6. Establishment and validation of the results in a nomogram
Nomogram is widely used as prognostic devices in oncology research. This approach can generate an individual probability of a specific clinical event via the integration of different determinant and prognostic variables [12–14]. In our study, we established a nomogram to evaluate the 1-year, 3-year, and 5-year OS probability of GC patients in the GEO cohort. We adopted Harrell’s concordance index (C-index) to evaluate the predictive power of the nomogram (combined model). The C-index is calculated using a 1,000 resampled boot method, whose values range from 0.5 to 1.0. In general, the C-index is less accurate at 0.50–0.70, moderately accurate at 0.71–0.90, and highly accurate above 0.90. The calibration of the nomogram was accomplished by drawing a comparison between the prediction probabilities and the observation probabilities. The closer the probabilities were to the reference line, the higher the consistency of the model. Simultaneously, single prognostic factors were utilized to construct the nomograms, and the prediction accuracy with the nomogram was compared by using C-index, ROC analysis, and decision curve analysis (DCA). DCA is a novel method for evaluating prognostic strategies, with the ability to visualize the clinical effectiveness of the nomogram [15]. All of the above methods were implemented in the R language.
Results
1. Identification of DEGs between GC and nontumorous tissues
To better illustrate our research, we have drawn an analysis process flow chart (Fig. 1). In the mRNA expression profile of GC patients (n=298), a total of 252 DEGs (LogFC > 2 or LogFC < −2, adjusted p < 0.001) were found in comparison with normal tissues (n=99). Among these DEGs, 163 genes were downregulated and 89 genes were overexpressed. Differentially expressed mRNA heatmaps and volcano maps are shown in S1 and S2 Figs.
Then, univariate Cox regression analysis was applied to explore the DEGs related to OS, and 130 genes (p < 0.001) were identified after primary filtration (Fig. 1). To further reduce the candidate genes, we conducted a Lasso-penalized Cox analysis. As a result, five genes met the screening criteria, which appeared more than 900 times in 1,000 screenings (S3 Fig.). Ultimately, we performed a stepwise multivariate Cox regression analysis, and four candidate genes were finally selected to establish the prognostic model. Among them, RNA binding protein, RBPMS2, and RGN were downregulated in tumor tissues, and the other two genes, PLEKHS1 and CT83, were overexpressed in tumor tissues.
2. The prognostic model shows good performance in risk stratification and ROC curve verification for GC patients
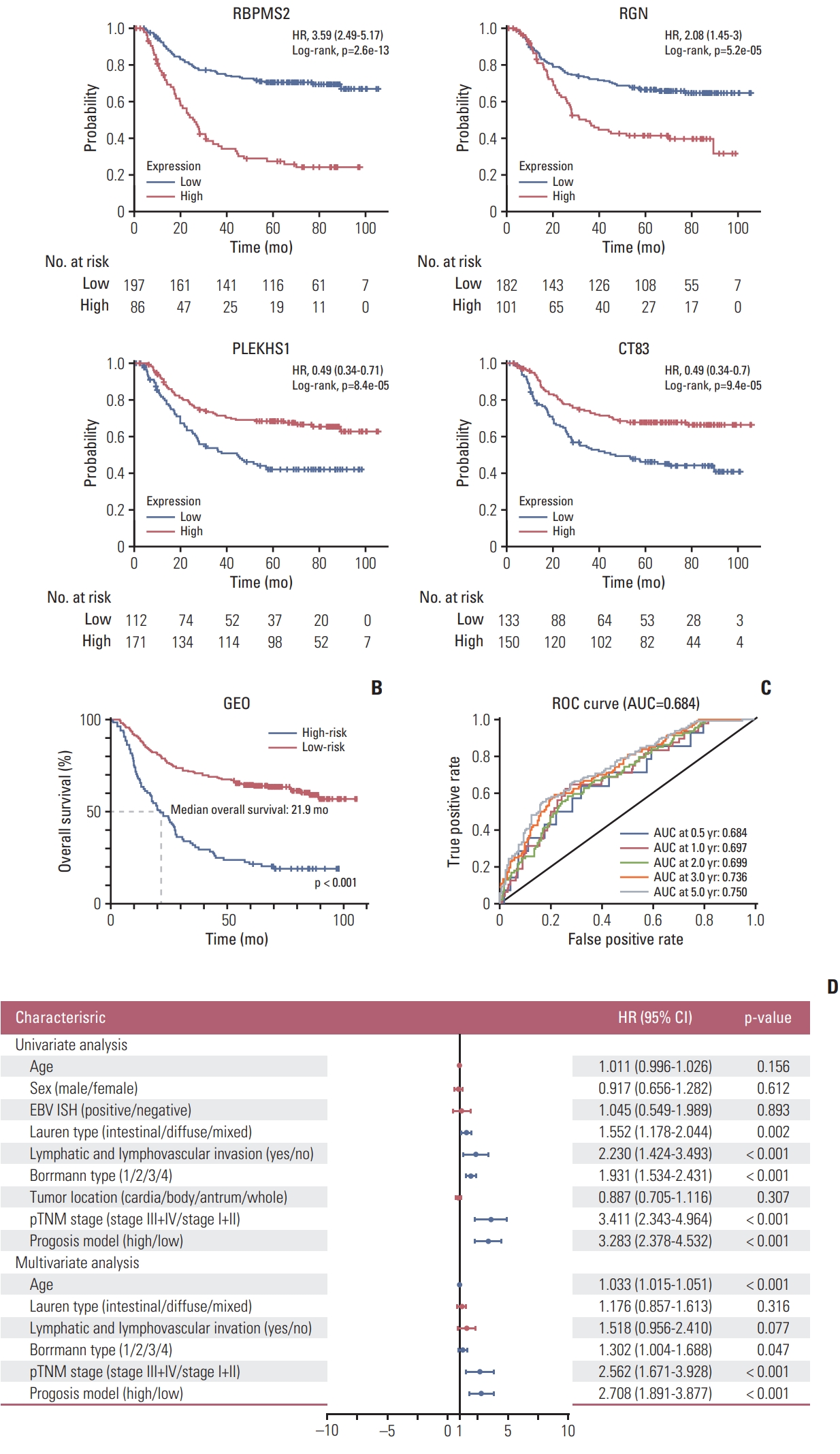
We built a predictive model using the four genes selected above and divided the GC patients into a low-risk group and a high-risk group according to the risk scores. The risk score=(expression level of RBPMS2×0.308)+(expression level of RGN×0.192)+(expression level of PLEKHS1×−0.197)+ (expression level of CT83×−0.190). RBPMS2 (hazard ratio [HR], 1.360; 95% confidence interval [CI], 1.172 to 1.579) and RGN (HR, 1.211; 95% CI, 1.010 to 1.453) showed positive coefficients, indicating that these two genes are high-risk factors in GS, while PLEKHS1 (HR, 0.821; 95% CI, 0.680 to 0.991) and CT83 (HR, 0.827; 95% CI, 0.694 to 0.985) showed negative coefficients, suggesting that their overexpression signified a longer OS. Verification in KM Plot supports the assumption (Fig, 2A), the high expression of RBPMS2 and RGN was negatively correlated with the patient’s prognosis, while the expression of PLEKHS1 and CT83 was opposite, and it was positively correlated with the patient’s prognosis. The optimal cutoff value of the risk score determined by X-tile software was 1.2 (S4 Fig.). As a result, 88 patients were classified into the high-risk group, and the other 210 patients were classified into the low-risk group. The Kaplan-Meier survival curves of the two risk groups showed significant differences (low-risk group vs. high-risk group: median OS, > 100 months vs. 21.9 months, respectively; p < 0.001) (Fig. 2B). ROC analysis confirmed the sensitivity and specificity of the prognostic model. The area under curves (AUCs) of the prognostic model were 0.684, 0.697, 0.699, 0.736, and 0.750 for 0.5-year, 1-year, 2-year, 3-year, and 5-year survival, respectively (Fig. 2C). In general, the prognostic model shows good performance in risk stratification for GC patients.
3. Univariate and multivariate Cox regression analysis proves that the prognostic model has good independence
We conducted univariate and multivariate Cox regression analyses on the diagnostic model to assess its independent predictive value with other conventional clinical factors in 298 GC patients from the GEO cohort. The univariate Cox regression showed that Lauren type (intestinal/diffuse/mixed), lymphatic and lymphovascular invasion (yes/no), Borrmann type (1/2/3/4), pTNM stage (stage III+IV/stage I+II), and prognostic model (high/low) were correlated with OS (Fig. 2D). However, age, sex, Epstein-Barr virus in situ hybridization status, and tumor location had no prognostic value in univariate Cox regression. Then, we performed multivariate Cox regression analysis on meaningful indicators in the results of univariate Cox regression analysis, and age was also included in the analysis. Finally, age, Borrmann type, pTNM stage, and prognostic model were shown to be independent predictors of OS (Fig. 2D).
4. The predictive model works well in the TCGA Asian population and is not effective in non-Asian populations
Validation of the prognostic model was conducted using TCGA database. In the TCGA global cohort, 110 patients were classified into the high-risk group, and the other 198 patients were classified into the low-risk group. Consistent with the result in the training set, patients in the low-risk group had a better prognosis than the higher-risk group (p=0.008) (Fig. 3A). However, the difference in OS is not as obvious as in the training set. Considering that GC has significant biological differences between different races, we further validated the prognostic model in TCGA Asian cohort and TCGA non-Asian cohort. In TCGA Asian cohort, a total of 23 patients were classified into the high-risk group, and 39 patients were classified into the low-risk group. The survival curves are highly consistent with the GEO cohort (p=0.002) (Fig. 3C). However, in the TCGA non-Asian cohort, the survival curve was similar to the TCGA global cohort, but it is not statistically significant (p=0.115) (Fig. 3E). Furthermore, ROC analysis confirmed the sensitivity and specificity of the prognostic model in the TCGA Asian cohort (AUC, 0.694), and AUCs for 0.5-year, 1-year, 2-year, 3-year, and 5-year survival were 0.694, 0.748, 0.741, 0.754, and 0.754 respectively (Fig. 3D). In the TCGA global cohort and TCGA non-Asian cohort, the AUCs are 0.593 and 0.567 (Fig. 3B and F), suggesting that the prognostic model has limited predictive power in these two cohorts.
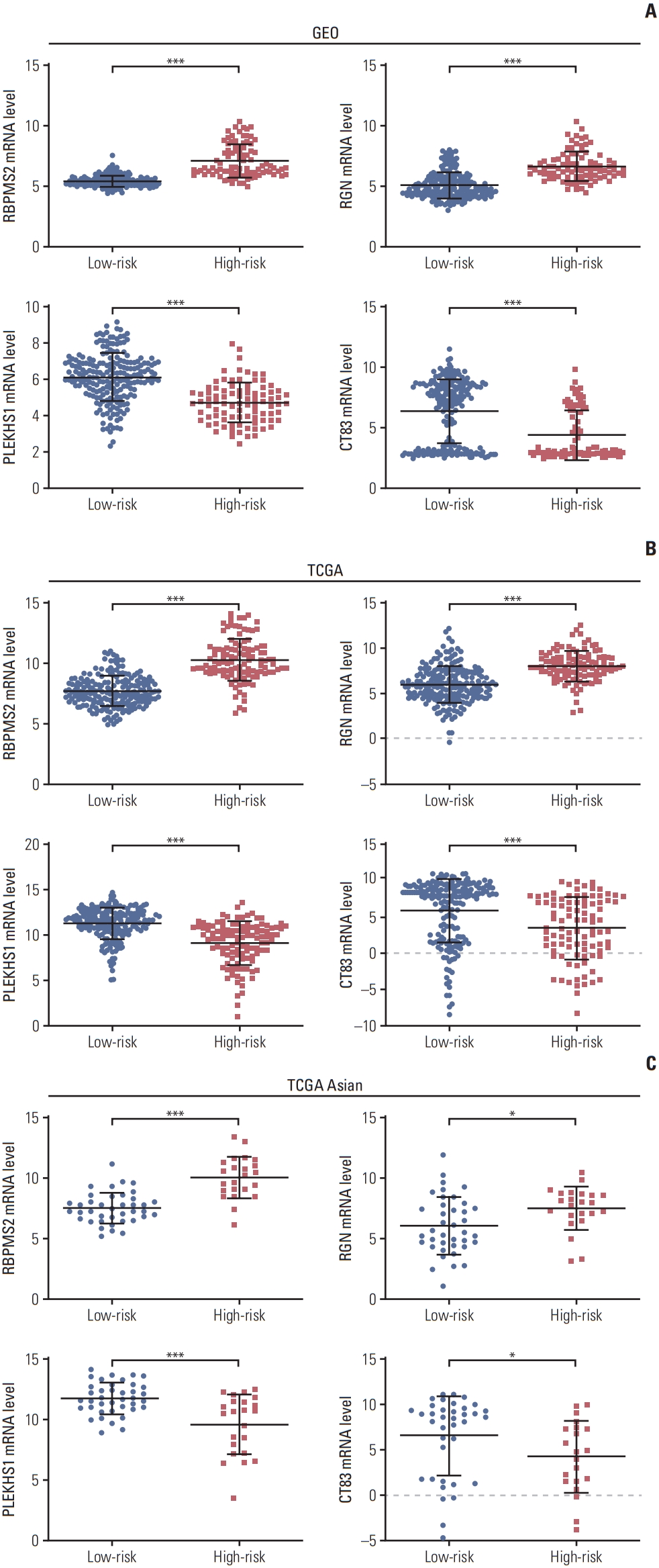
5. Verification of the expression levels of the four genes in TCGA datasets
In the GEO cohort, RBPMS2 and RGN were overexpressed, while PLEKHS1 and CT83 were downregulated in the high-risk group (Fig. 4A). To further verify the accuracy of this result, we compared the expression of these four genes in the TCGA global cohort (Fig. 4B) and the TCGA Asian cohort (Fig. 4C). The results were similar to the GEO cohort.
6. Establishment and validation of the nomogram
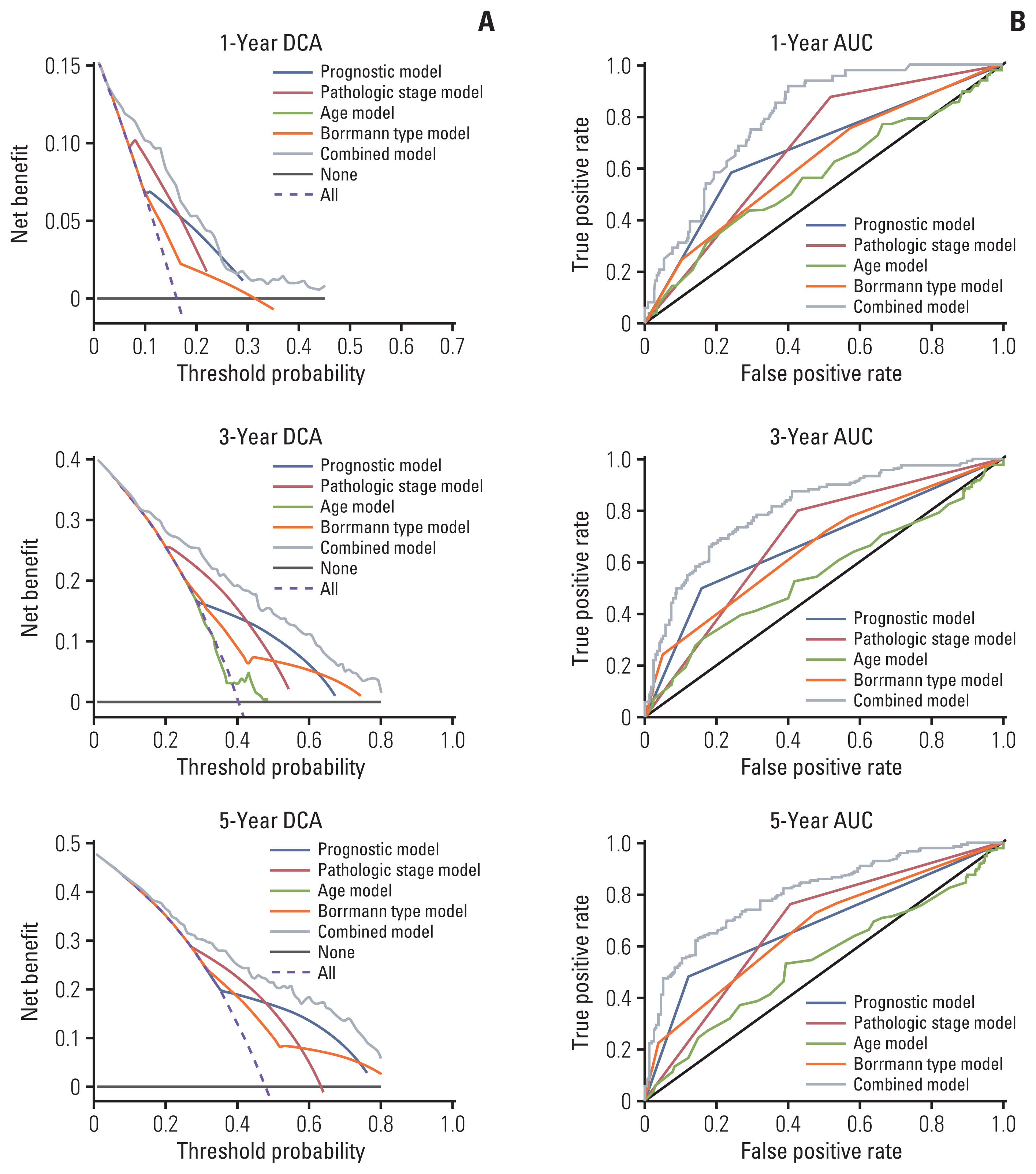
To establish a more convenient and accurate survival prediction method for patients with GC, we built a nomogram using the results of multivariate Cox regression analysis. The OS-related factors include age, pathologic stage (III+IV/I+II), Borrmann type (1/2/3/4), and prognostic model (high/low) (Fig. 5A). We used calibration plots to verify the nomogram (Fig. 5B). The C-index for the nomogram (combined model) was 0.74 (95% CI, 0.71 to 0.78), which was significantly higher than in other models. (Table 1). ROC analysis and DCA were performed to compare the prediction accuracy between single clinical factors and the nomogram. The DCA curve showed that the nomogram had the strongest predictive power and accuracy among several predictive models (Fig. 6A). The results of the ROC analysis show that the AUC of the nomogram was the largest (Fig. 6B).
Discussion
GC remains one of the most commonly diagnosed malignancies and leads to cancer death worldwide, especially in Asia [1]. There is tremendous pressure on developing techniques for the prevention and treatment of GC. Survival prediction affects the choices of further treatment options. Traditional survival prediction methods using pathological information and serum tumor marker levels as the main means have played an important role in clinical practice over the past few decades [16]. However, with the advent of the era of precision therapy, it is gradually becoming difficult for this predictive approach to meet clinical needs. GC has significant biological and epidemiological differences between Asian and non-Asian populations; it is difficult to find a one-size-fits-all approach for all patients. Therefore, in order to improve the efficacy of GC and reduce mortality, there is an urgent need for different survival prediction methods for patients of different races.
The GSE66229 dataset contains microarray profiles of gastric tumors from Asian patients. Previously, Cristescu et al. [8] used multi-omics data (including GSE66229) to classify GC into MSS/TP53+, MSS/TP53−, MSS/EMT, and MSI molecular types and described the molecular characteristics of GC at the genetic level. Moreover, Zhang et al. [17] used the GSE66229 dataset to investigate the significance of crosstalk between long non-coding RNA and mRNA in GC. In our study, we used the GEO dataset GSE66229 to select four genes, RBPMS2, RGN, PLEKHS1, and CT83, to establish a diagnostic model. The coefficient and hazard ratio showed that RBPMS2 and RGN are high-risk factors in GC, while the overexpression of PLEKHS1 and CT83 signify a longer OS. The AUCs of the ROC curves were 0.684, 0.697, 0.699, 0.736, and 0.750 for 0.5-year, 1-year, 2-year, 3-year, and 5-year survival, respectively. This model performed well in survival prediction. In this model, patients can be assigned to low-risk or high-risk groups based on risk scores, providing a basis for further precision treatment. Diagnostic models can also be used to guide postoperative adjuvant chemotherapy. For patients in the high-risk group, a more potent combination chemotherapy regimen should be used to inhibit and destroy tumor cells. Moreover, in response to high-risk groups, a more detailed review strategy can be developed to enable early detection of recurrent cases. We also demonstrated the independence of the prognostic model with other clinical data in GC.
Next, we validated the diagnostic model in the TCGA global cohort. Considering that GC has significant biological differences between different races [18], we further validated it in the TCGA Asian cohort and the TCGA non-Asian cohort. And the results were finally verified in the TCGA Asian cohort, demonstrating that the prognostic model based on the GEO dataset from Korea is capable of dividing Asian GC patients into a high-risk group or a low-risk group and predicting OS. However, the predictive power was limited in the non-Asian population. Similar ethnic differences have also been found in epidemiological investigations in the United States. The incidence of GC in Japanese immigrants to Hawaii was higher than that of local residents in Hawaii [19]. Interethnic differences can, therefore, play an important role in the development and progression of GC.
The protein encoded by RBPMS2 is a member of the RNA recognition motif-containing protein family and plays an important role in regulating the development and dedifferentiation of digestive smooth muscle cells [20]. Previous research in chick embryos showed that RBPMS2 was expressed in the early stages of the visceral smooth muscle cell and gradually decreased with the maturity of smooth muscle cells. In differentiated primary cultured smooth muscle cells, ectopic expression of RBPMS2 upregulates cell proliferation rates and inhibits cell contractile function [21]. Aberrant elevated expression of RBPMS2 can be specifically observed in gastrointestinal mesenchymal neoplasm and digestive myopathy syndrome, demonstrating that the regulated expression of RBPMS2 is important for the proper development and differentiation of visceral smooth muscle cells [21,22] In our research, the expression of RBPMS2 in the high-risk group was significantly higher than that in the low-risk group. This finding suggests that cases in the high-risk group are more likely to invade the muscular layer and cause abnormal functioning of smooth muscle cells. RBPMS2 can be defined as a novel marker of visceral smooth muscle remodeling for characterizing smooth muscle layer invasion in gastrointestinal cancer.
RGN is a protein-coding gene. Previous studies on breast cancer and lung cancer have demonstrated that it may play a crucial role in maintaining intracellular Ca2+ homeostasis, suppressing cell proliferation, inhibiting oncogene expression, and increasing tumor suppressor gene expression [23,24]. Our research also found that RGN was downregulated in tumor tissues compared with adjacent nontumor tissues. However, in GC patients, RGN expression is higher in the high-risk group than in the low-risk group. To further validate the relationship between RGN expression levels and survival time, we validated the GEO data using the KmPlot website and found that the results regarding RGN expression in lung cancer and breast cancer are consistent with those reported in other works in the literature, but the results were reversed in GC. The RGN high-expression group has a shorter survival period, which coincides with our findings.
At present, there are few studies on PLEKHS1, and the prognostic value of PLEKHS1 in GC has not been verified in previous studies. Mutations in non-coding regions of PLEKHS1 were found in cancer patients according to a genome-wide analysis, which may be related to the degree of tumor malignancy [25]. Our study found that PLEKHS1 is a protective factor in patients with GC and that its expression is higher in the low-risk group, as in previous reports [26].
CT83, also known as KK-LC-1, is a gene that encodes a member of the cancer-testis antigenic protein family and is only expressed in malignant tumor tissue and testicular germ cells [27–29]. Based on this feature, Marcinkowski et al. [27]believe that CT83 may become an attractive target antigen for chimeric antigen receptor T-cell immunotherapy (CAR-T). In our results, the expression level of CT83 in the low-risk group was significantly higher than that in the high-risk group. We speculate that CT83 may be related to the body’s anti-tumor response. In normal tissues, the expression of CT83 is at an extremely low level. When tumors occur, the expression level of CT83 may be related to the immune response of the body against tumors. The higher the expression levels of CT83, the stronger the body’s ability to fight tumors, so the better the prognosis. Moreover, in the study of the early diagnosis of GC, Futawatari et al. [30] found that high CT83 expression rates can be frequently detected in the early stage of GC. Therefore, CT83 can be used as a potential marker for the early diagnosis and treatment of GC.
The current risk assessment for patients with GC is based primarily on the TNM staging system [14]. This anatomical-based approach has played an important role in clinical practice over the past few decades, but it is still difficult to explain why patients with the same staging receive different prognoses. With the advent of the era of precision therapy, doctors need more accurate methods for patient risk analysis. Our nomogram combines genetic information with GC clinical data, to make the prediction ability 15.6% higher than that only focus on the pathologic stage (C-index, 0.74 vs. 0.64; p < 0.001). Another advantage of our predictive model is that it only needs to detect the expression levels of the four genes rather than somatic mutations in patients, which can be achieved by some simple and economical methods, greatly reducing the cost of sequencing.
However, there are still some limitations to this research. Our nomograms were not externally validated in the TCGA database because of the lack of data on Bormann type. In the future, we will detect the expression levels of the four genes in more clinical samples to verify and improve this prediction model.
To sum up, our four-gene–related prognostic model and the nomogram are reliable tools for predicting the OS of Asian patients. However, the predictive power is limited in the non-Asian population. Our nomogram will help stratify Asian patients with GC more accurately and promote more precise treatment for all of them.