Introduction
In Korea, colorectal cancer (CRC) is the third most prevalent cancer and the most common cancer among women aged 65 years and older with estimated 54.9 new cases, per 100,000 men and women, per year and responsible for 10.7% of cancer deaths [1]. Even though ongoing efforts to establish an early preventive screening programs have reduced the incidence of advanced stage disease, and thus led to the improvement in 5-year overall survival rates up to 70.6% [2]. However, the progress in the development of therapeutic options for metastatic CRC (mCRC) was dismal until recent discoveries in molecular-based therapies.
Over the past decade, large-scale sequencing studies have advanced our understandings of molecular diversity among mCRC patients and uncovered potentially actionable genes across subgroups of patients with distinct molecular signatures including WNT, RAS-MAPK, PI3K, TGF-β, p53, and DNA mismatch repair pathway [3]. Next-generation sequencing (NGS) can facilitate precision medicine approaches by identifying actionable somatic events in tumor samples and reveal associations with sensitivity or resistance that can inform the development and implementation of targeted therapeutics and aid in the design of clinical trials to validate findings and actionability [2,4-6].
In June 2017, The K-MASTER cancer precision medicine diagnosis and treatment enterprise (K-MASTER project) was initiated as a nationwide precision medicine oncology clinical trial platform, which used NGS assay to screen the actionable mutations in 10,000 Korean patients with refractory solid tumors and assigned the patients to matched clinical trials. As of January 2020, 52 sites and 4,390 cancer patients have participated. Under the K-MASTER program, we investigate the current mCRC treatment landscape according to molecular genomic alterations and discuss potentially actionable genes currently being investigated in clinical trials that may provide further progress in this battle against this malicious disease. By analyzing the gap between potential theoretical possibilities and current treatment practice, we can try to find a way to realize the unmet need of mCRC patients.
Materials and Methods
1. Patients samples
Pathologically confirmed mCRC patients (n=994) have been referred from 54 institutions located all over South Korea between June 2017 and January 2020 under K-MASTER project (S1 Table). Specimens were prepared from formalin-fixed and paraffin-embedded tumor tissue (n=842) or blood if tumor tissue was not available (n=152). The most recent archival tissue, either from the primary tumor or a metastatic site, was reviewed for tumor cellularity. Details of the genomic DNA extraction are described in Supplementary Method.
2. Targeted sequencing and bioinformatics
Targeted sequencing was performed using three NGS platforms such as SNUH FIRST Cancer Panel [2], K-MASTER Cancer Panel [4], and Axen Cancer Panel 1. SNUH FIRST panel v3.01 was performed with exons of 183 cancer-related genes including microsatellite status with five microsatellite markers (D2S123, D5S346, D17S250, BAT25, and BAT26) and K-MASTER cancer panel v1.1 included the whole exomes of 409 cancer-related genes, the intronic regions of 23 genes and three fusion genes. Axen Cancer Panel 1, a liquid biopsy panel, was utilized which panel included the exomes of 88 cancer-related genes and the intronic regions of three genes (S2 Table). Detailed methods for targeted sequencing and variant annotation can be found in the Supplementary Method. Targeted sequencing generated approximately 100 Mb per sample with an average of 95% samples on target. Sequences of all samples achieved a mean depth was more than 650× of FIRST and K-MASTER panel and 2580x of Axen panel.
The molecular alterations as single nucleotide variants, insertions and deletions (indels), copy number alterations, or structural variants with the potential to affect clinical decisions or impact the way patients are enrolled in clinical trials. Detected DNA alterations were annotated as pathogenic variants as the case of “likely-pathogenic” or “pathogenic” in the COSMIC and ClinVar and “likely-oncogenic” or “oncogenic” in the OncoKB database.
Results
1. Pathogenic variants in mCRC
This analysis included 994 mCRC patients, with 1,564 clinically meaningful pathogenic variants in 71 mutated genes (Table 1, S3 Table). On average, there are 1.5 clinically meaningful pathogenic variants per patient.
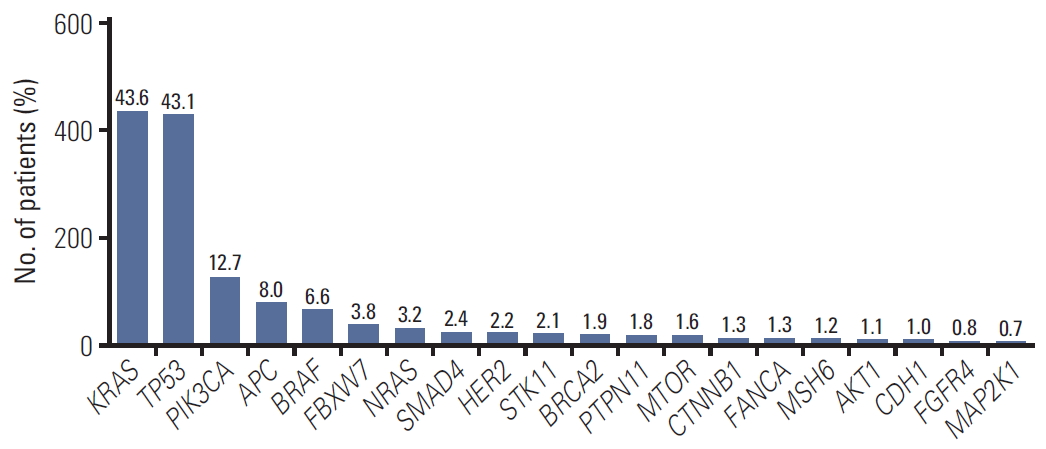
The top 20 frequent genes are as follows: KRAS (n=443, 43.6%), TP53 (n=428, 43.1%), PIK3CA (n=126, 12.7%), APC (n=80, 8.0%), BRAF (n=66, 6.6%), FBXW7 (n=38, 3.8%), NRAS (n=32, 3.2%), SMAD4 (n=24, 2.4%), HER2 (n=22, 2.2%), STK11 (n=21, 2.1%), BRCA2 (n=19, 1.9%), PTPN11 (n=18, 1.8%), MTOR (n=16, 0.6%), CTNNB1 (n=13, 1.3%), FANCA (n=13, 1.3%), MSH6 (n=12, 1.2%), AKT1 (n=11, 1.1%), CDH1 (n=10, 1.0%), FGFR4 (n=8, 0.8%), and MAP2K1 (n=7, 0.7%) (Fig. 1, S4 Fig.).
2. Current treatment landscape of mCRC
1) Targeting anti-EGFR pathway
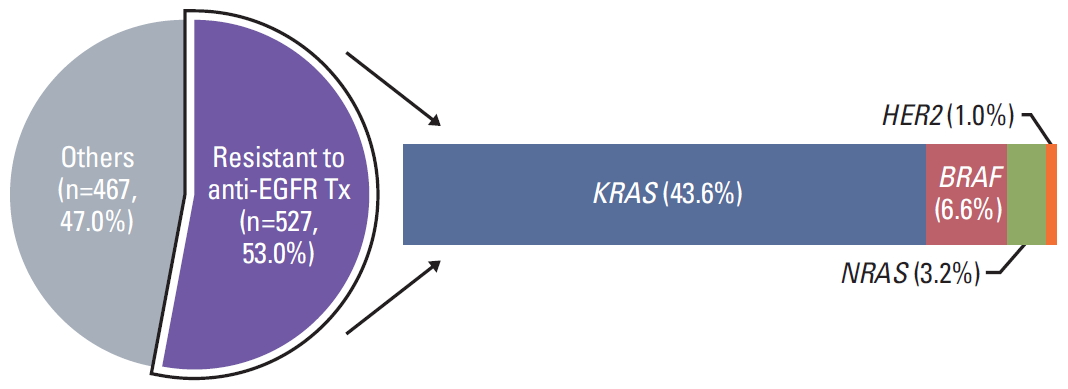
Anti-epidermal growth factor receptor–directed monoclonal antibodies (anti-EGFR mAbs) were the first targeted therapies for mCRC which were active both as single agents and in combination with cytotoxic chemotherapy [5,6]. Out of 994 patients, 467 patients (47.0%) were able to get anti-EGFR therapy with wild-type RAS. The other 527 patients (53.0%) were having pathogenic variants in KRAS (n=433, 43.6%), BRAF (n=66, 6.6%), NRAS (n=32, 3.2%), or HER2 amplification (n=10, 1.0%) which were known to be resistant to anti-EGFR therapy and avoided as an ineffective drug for treatment decision (Fig. 2) [7,8] . Fourteen patients have concurrent mutations related with anti-EGFR therapy resistant genes; seven patients with KRAS plus BRAF mutation, three patients with KRAS plus NRAS mutation, two patients with NRAS plus BRAF mutation, and two patients with KRAS mutation plus HER2 amplification.
2) Targetable genes with approved indications
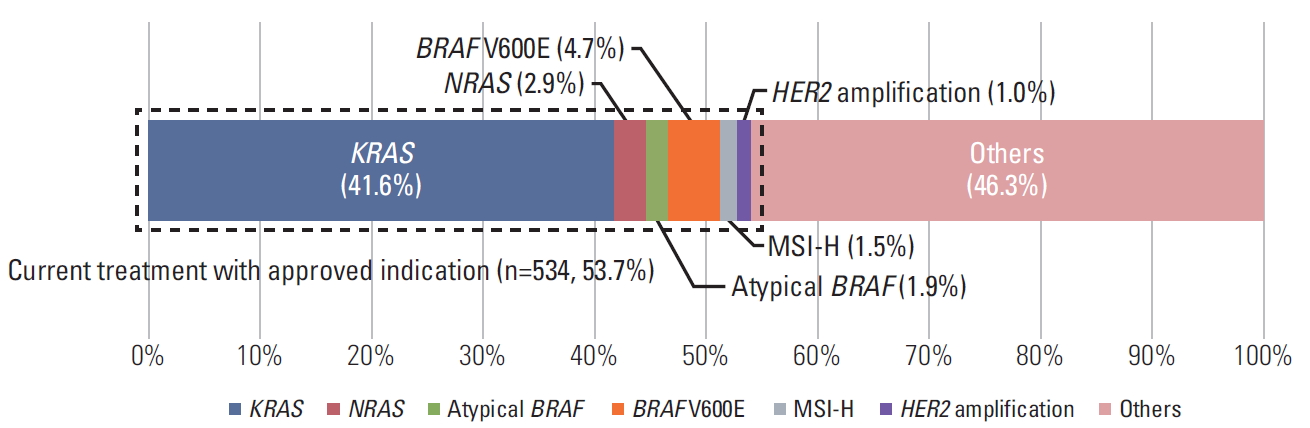
Beyond anti-EGFR therapy with wild-type RAS, there are several biomarkers that can help using targeted therapy or immunotherapy. For patients with BRAF V600E mutated mCRC, the combination of encorafenib and cetuximab or panitumumab was recommended to second- or third-line mCRC treatment [8]. Dual blockade of anti-HER2 agents such as trastuzumab plus pertuzumab [9] or trastuzumab plus lapatinib [10] could be incorporated in HER2-amplified mCRC after progression of second-line cytotoxic chemotherapy. Pembrolizumab [11] and nivolumab [12] (or ipilimumab combination) [13] were approved for subsequent-line treatment of mismatch repair genes (MMR includes MLH1, MSH2, MSH6, PMS2, and EPCAM) deficient mismatch repair (dMMR)/microsatellite instability (MSI)–high cancers based on high response rates and encouraging clinical outcomes. In our study, there were mCRC patients with BRAF V600E mutation (n=47, 4.7%), dMMR/MSI-high (n=15, 1.5%, one patient had two pathogenic variants with MSH2 deletion and MSH6 T1102fs simultaneously), HER2 amplification (n=10, 1.0%) which meant additional 72 patients (7.2%) could have had an opportunity to treat with promising drugs (Fig. 3). While 53.7% of all patients with mCRC can receive the customized treatment based on the type of pathogenic mutations, the remaining 46.3% of patients have unmet need of precision medicine approach.
3) Candidates for immune checkpoint inhibitors
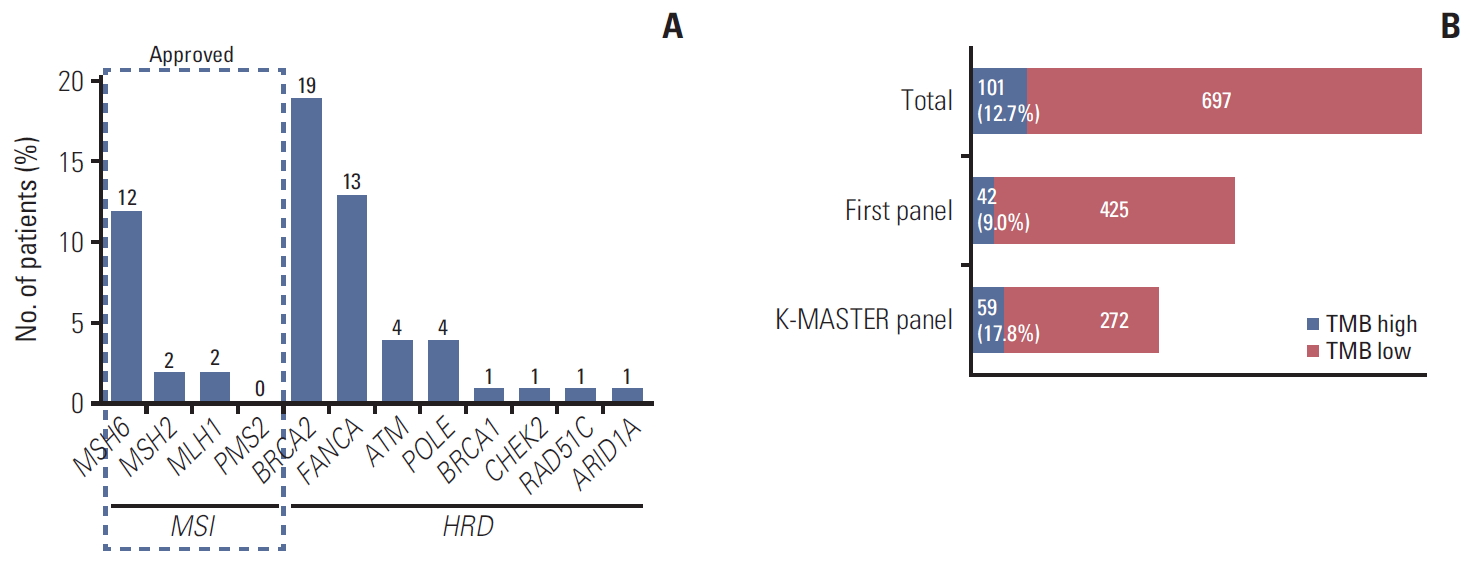
A small subset of the microsatellite stable tumors exhibited high tumor mutational burden (TMB) and DNA damage and repair (DDR) defected cancer patients responded immune checkpoint inhibitors [14]. In our study, 42 patients (4.2%) have pathogenic variants in defected DDR pathway genes (homologous recombination deficiency genes, HRD genes) such as BRCA1/2, CHEK1/2, FANCA, ATM, POLE, RAD51B/C/D, RAD54L, ARID1A, BARD1, BRIP1, CDK1/2, PALB2, etc. (Fig. 4A). Each patient had one pathogenic variant except one who had three pathogenic variants at the same time (BRCA2, FANCA, ATM).
Out of 798 patients analyzed TMB with K-MASTER panel (n=331) or FIRST panel (n=467). TMB cut-off value defined as > 16 per 106 base pair in K-MASTER panel [4] and as > 13 per 106 base pair in FIRST panel [15] which were 59 patients (17.8%) and 42 patients (9.0%) irrespectively. Although the definition of TMB is different for both panel, a number of patients (n=101, 12.7%) might be treated with immune checkpoint inhibitor approximately (Fig. 4B) [16].
4) Actionable genes under ongoing clinical trials
There are several ongoing trials targeting PIK3CA with RAS/RAF/PTEN/PIK3CA blockade, KRAS G12C with AMG 510, atypical BRAF with BRAF/MEK blockade, and HER2 mutation with anti-HER2 agents for mCRC patients. If the patients can participate in these clinical trials, total 180 patients (18.1%) who have PIK3CA (n=126, 12.7%), KRAS G12C (n=30, 3.0%), atypical BRAF (n=17, 1.7%), and HER2 mutations (n=12, 1.2%, one patient has concurrent mutations with HER2 amplification) can get an opportunity to treat with promising drugs.
If we extend these pathogenic variants to all solid tumors beyond mCRC, clinical trials with high probability of success are underway with actionable genes such as NRAS (n=32, 3.2%), MTOR (n=16, 1.6%), AKT1 E17K (n=9, 0.9%), MAP2K1 (n=7, 0.7%), FGFR1 amplification (n=6, 0,6%), CDKN2A (n=5, 0.5%), FLT3 amplification (n=5, 0.5%), EGFR amplification (n=5, 0.5%), HRAS (n=4, 0.4%), CDK4 amplification (n=3, 0.3%), MET amplification (n=3, 0.3%), PTEN (n=3, 0.3%), RET (n=3, 0.3%), TP53 E285K (n=3, 0.3%), IDH1 (n=2, 0.2%), MDM2 amplification (n=2, 0.2%), KRAS G13V (n=1, 0.1%), KIT (n=1, 0.1%), NF1 R1276Q (n=1, 0.1%), TSC2 (n=1, 0.1%), and HRD genes (n=42). Putting all these rare mutations together, these 137 patients (13.8%) might be able to challenge to get therapeutic opportunities.
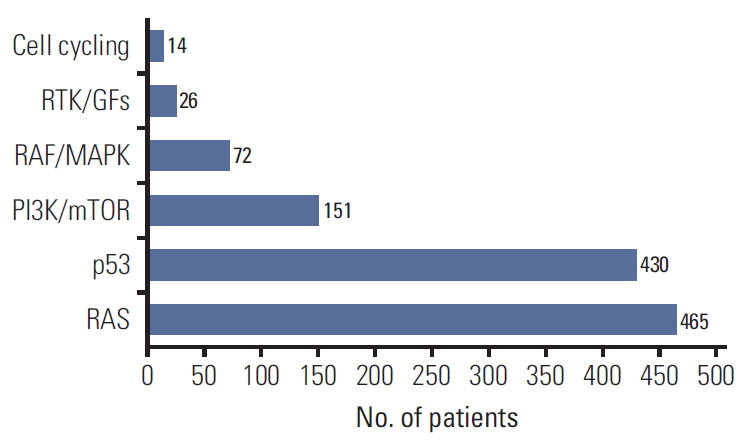
We classified pathogenic variants by critical signaling pathways (Table 2). Total 465 patients (46.8%) have pathogenic variants in RAS pathway, 430 patients (43.3%) in p53 pathway, 151 patients (15.2%) in PI3K/mTOR pathway, 72 patients (7.2%) in RAF/MAPK pathway, 26 patients (26.1%) in receptor tyrosine kinase/growth factor pathway, and 14 patients (14.1%) in cell cycling pathway (Fig. 5).
There are several fusion mutations that indeterminant significance in CRC but significant in other solid tumors; ALK-EML4 fusion genes (n=2, 0.2%) and RET-NCOA4 fusion genes (n=4, 0.4%) (S5 Fig.) are significant in non–small cell lung cancer. NTRK fusion genes are very difficult to find the right patients in the daily practice setting but it is known that the treatment response rate is very high if drugs targeting pathogenic variants are incorporated [17-20].
Discussion
Cancer treatment is moving to a new paradigm where the molecular characteristics of the tumor are used to inform treatment decision. In recent decades, people investigated the genetic basis of CRC and large-scale sequencing studies discovered key pathways such as WNT, RAS/MAPK, PI3K, TGF-β, p53, and dMMR. Although our understanding of genomic alteration of mCRC has rapidly improved, we still do not fully understand the heterogenous properties of CRC and do not provide the personalized treatments based on individual molecular characteristics.
In this study, we investigated mutations within 409 selected oncogenes and tumor suppressor genes and found 71 genes with 1,564 pathogenic variants from 994 mCRC patient samples. Overall, our results showed that our population had a similar mutation profiles to populations that reported in other studies [21-25], with alterations in driver genes, such as KRAS, TP53, PIK3CA, APC, BRAF, and FBXW7.
In order to overcome a resistance mechanism to anti-EGFR therapy, the innovative challenge has been initiated in mCRC harboring identified acquired targets such as BRAF mutation, HER2 amplification, and c-MET amplification by means of NGS [8,26]. Nonetheless, we are currently only able to apply anti-EGFR therapy (n=527, 53.0%) as a negative predictive biomarker under the conditions of Korean regulatory licenses. The rare mutations such as BRAF V600E (n=47, 4.7%), MSI-high (n=15, 1.5%), HER2 amplification (n=10, 1.0%) are rising targets with novel agents which neither approved nor reimbursed in Korea so these population could not be helped determining treatment at this moment.
Although there is a difference in the cut-off value between two panels, a significant number of TMB high mCRC patients should be considered as a target population with immune checkpoint inhibitors. Even rarer genetic alterations are also considered as actionable targets. For example, 30 patients with KRAS G12C could be targeted with AMG 510 as a first-in-class KRAS G12C inhibitor. EML4-ALK fusion (n=2) and RET-NCOA4 fusion (n=4) occurred in less than 1% of cases which were not classified as a pathogenic variant due to no evidence in CRC but could be the targets. Clinical trials should be conducted with various promising drugs for mCRC patients in order to expand therapeutic opportunities. For example, 180 patients (18.1%) with KRAS G12C/atypical BRAF/PIK3CA, HER2 mutations could be participated in ongoing trials within mCRC and 137 patients (13.8%) could take part in clinical trials in all solid tumors with novel variants. If all mCRC patients with potentially actionable variants would have challenged to get therapeutic opportunities, additional 294 patients (29.6%) would have tried to enroll the clinical trial (Fig. 6).
Under K-MASTER project, a nationwide cancer genome screening project in Korea, this study aimed to identify key alterations in mCRC that may represent important targets for novel drugs using customized cancer panels at the nationwide level. K-MASTER project with customized cancer panel can be accomplished in a realistic timeframe for use in dayto-day clinical care and could be used to expand treatment options for mCRC patients. Using efficient genomic screening systems, relatively minor cancer genome alterations are necessary for the successful development of targeted therapies under clinical trials.
This mCRC study assessing the feasibility of precision oncology for mCRC patients in real-world setting found that about 52.8% of patients will able to treat with targeted therapies and 29.6% of patients could be candidate of clinical trials with innovative drugs. It is believed that a precise medical approach can contribute to broadening the treatment opportunities for mCRC patients.