INTRODUCTION
Gastric cancer is one of the most common cancers in Korea and all of Asia (1) despite of the tendency for its reduced incidence and mortality. There is no gold standard combination chemotherapeutic regimen that is effective therapy against gastric cancer. Therefore, more effective anticancer agents need to be developed.
DNA topoisomerases are enzymes that can alter the topology of DNA by transiently breaking one or two strands of DNA, passing a single- or double-stranded DNA through the break and finally resealing the breaks. These enzymes are involved in a number of crucial cellular processes, including replication, transcription and recombination. Therefore, they are considered as important therapeutic targets for anticancer chemotherapy. Topoisomerase I (topo-I) and topoisomerase II (topo-II) inhibitors have recently been introduced as promising anticancer agents because of their high sensitivity and strong anticancer activity for treating various cancers (2). In particular, irinotecan (CPT-11), a semisynthetic derivative of camptothecin (CPT), showed strong anticancer activity against leukemia, lymphoma (3), small cell lung (4), non-small lung (5), and colorectal cancer (6).
7-{2-(N-isopropylamino)ethyl}-(20S)-camptothecin, or belotecan (Camtobell®, CKD602) (Fig. 1) is a recently synthesized analogue of camptothecin (CPT) (8) for which a water-solubilizing group was introduced at position 7 of the B-ring. A hydroxyl group (-OH) was also introduced at position 20 of the E-ring, which enhances the anticancer activity of this agent (7). Belotecan has some differences from the other camptothecin-derived anticancer drugs. It has higher water-solubility than the other camptothecin-derived anticancer drugs (>5 mg/kg) (7). The anticancer activity of belotecan stems from its inhibitory effect on replication of single strains of DNA, which is induced by the formation of a stable complex with DNA topo-I complex during the replication of DNA. The efficacy of belotecan has been evaluated by several in vitro assays, including a cell cytotoxicity assay and a cleavable complex assay (7). The anticancer activity was demonstrated in various cancer cell lines, including colon cancer (HT-29, WIDR, CX-1), lung cancer (LX-1), breast cancer (MX-1) and gastric cancer (SNU-5). The result from in vivo study also confirmed the anti-cancer effect of belotecan in SKOV-3, an ovarian cancer xenograft and L1210, a leukemia model (7). In a phase I study, the DLT (dose-limiting toxicity) of belotecan was demonstrated by the neutropenia and thrombocytopenia, but not by diarrhea, which is the prototype toxicity of CPT-11 (8). A phase II study showed that belotecan has significant activity against ovarian cancer (overall response: 45%) and small cell lung cancer (overall response: 45%) with tolerable toxicity (9).
The topo-I inhibitors that have been developed before belotecan include camptothecin, topotecan and irinotecan (CPT-11) (2-6). Though their high anticancer effect was established in case in which they were applied as single agents, their synergisms with cisplatin (CDDP), carboplatin, etoposide and 5-fluorouracil (5-FU) were also established via in vitro, ex vivo and clinical studies (10-12).
The synergisms of the recently developed topo-I inhibitors (TAS-103, NB-506 and SN-38) with other drugs has also been established (10, 11). However, there have been no studies done about the interaction between belotecan and other drugs and their mechanisms of interaction in gastric cancer.
Therefore, we conducted this study to demonstrate whether belotecan and cisplatin are synergistic and to elucidate the mechanisms that are responsible for the synergism between these two drugs in gastric cancer cells.
MATERIALS AND METHODS
1) Drugs and chemicals
Belotecan (Camptobell®, CKD602) was provided by Chong Kun Dang Co. Ltd. (Seoul, Korea). Cisplatin (CDDP, 10 mg/20 ml) was bought from Korean United Co. Ltd. (Seoul, Korea). The drugs were dissolved with distilled water just before their administration on each day of treatment and they were diluted to their final concentrations with using RPMI 1640 medium. [methyl-14C]Thymidine (52 mCi/mmol) was obtained from NEN Life Science Products, Inc. (Boston, MA); and TPA was obtained from Sigma-Aldrich Korea Ltd. (Korea).
2) Statistical analysis and graph
All statistics were calculated with Excel and the SPSS 11.0 for Windows software package. All graphs were made with Sigma Plot 8.0 (Jandel Scientific).
3) Cell line and culture
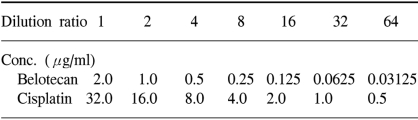
The human gastric cancer cell lines SNU-5, SNU-16 and SNU-601, which were originally established at Seoul National University College of Medicine, were donated by the Korean Cancer Research Resources Cell Bank. The SNU-5 and SNU-16 cells were grown as floating cultures and the SNU-601 cells were grown as attached cultures in RPMI 1640 medium (GIBCO, Grand Island, NY) supplemented with 10% v/v heat-inactivated fetal bovine serum (Sigma Chemical Co. St. Louis, MO) and gentamycin (100 µg/ml) in a humidified atmosphere of 5% CO2 in air at 37℃. The cells were routinely harvested by trypsinization and then they were diluted with the RPMI medium to the appropriate concentrations (Table 1). The cell sizes and numbers were measured by a Hemocytometer (Neubauer improved, Marienfeld. Germany).
4) Growth inhibition assay
We used the tetrazolium-dye (MTT) assay as described previously to evaluate the growth inhibitory effects of the cytotoxic agents (7). In brief, 50 µl aliquots of an exponentially growing cell suspension (1×106 cells/ml) were seeded onto 96-well microtiter plates (Nuclon, Denmark); the cells were incubated for 12 hours, after which time 150 µl aliquots of the drug solutions at various concentrations were added (Table 1). Following exposure to drugs, 50 µl of 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) solution (dissolved in DPBS (2 mg/ml)) were added to each well, and the plates were incubated at 37℃ for another 4 hours. After centrifugation of the plates at 4,500×g for 10 minutes, the medium was aspirated from each well as completely as possible, and 200 µl dimethylsulfoxide (DMSO) (Sigma) were added to each well to dissolve the formazan crystals. The absorbance was measured at 564 nm using Softmax ELISA (enzyme-linked immunosorbent assay analysis program) on a computer interfaced with a Thermomax microplate reader (DMC TMX BIO-RED; Molecular Devices). The wells containing only RPMI 1640-fetal bovine serum without cells were used as negative controls, and the wells containing media and cells without belotecan were used as positive controls. The IC50 was defined as the concentration that reduced the absorbance in each test by 50%. The absorbance was calculated as (mean absorbance of six wells containing drug - the absorbance of six control wells)/(mean absorbance of six drug-free wells - the absorbance of six control wells)×100. The IC50 value with 24 hours exposure was used as the representative value of the drugs. Each experiment was performed using six replicate wells for each drug concentration and each experiment was carried out independently three times.
5) Analysis of combination effects
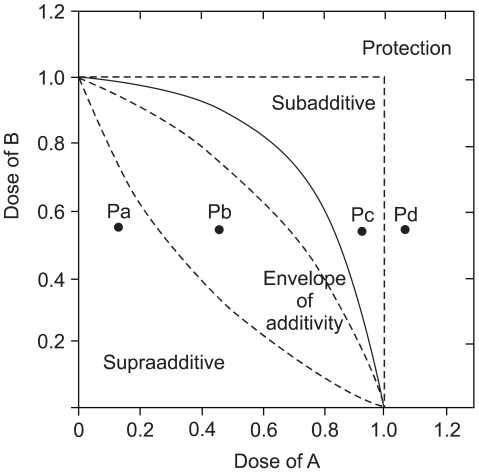
On the basis of the growth inhibition curve for each single drug, we analyzed the effects of the drug combination with using the isobologram method of Steel and Peckham. Three isoeffect curves (mode I, IIA and IIB) were drawn as follows (12) (Fig. 2). When the dose-response curve of drug A follows first-order kinetics, Mode IIb line will be identical to Mode I line and vice versa. When both drugs follow first-order kinetics, all three isoeffect lines will converge to make a straight line connecting 1.0 of the ordinate and abscissa. The total area enclosed by these 3 lines conceivably represents the additive response or an "envelope of additivity" (Fig. 2). A single dose-response curve can be drawn with the combinations of the graded doses of drug A and a chosen dose of drug B. When the experimental IC50 concentration of this combination falls left of the envelope (Fig. 2. Point Pa), then the 2 drugs have supraadditive interaction. When the experimental data point is within the envelope, then this combination is considered to be noninteractive (additive) (Fig. 2. Point Pb). When this point is in the area to the right of the envelope, but within the square produced by 1.0 of the ordinate and abscissa, then the drugs have subadditive interaction (Fig. 2. Point Pc). When the point is outside the square, then both drugs are considered to be mutually protective (Fig. 2. Point Pd). We chose to present a representative isobologram after confirmation that in repeated experiments, the individual combination data points were reproducibly in the same area of supraadditivity, additivity, or subadditivity irrespective of the envelope being slim or obese.
6) Alkaline elution assay
The exponentially growing gastric cell lines were radiolabeled by incubation with [methyl-14C]thymidine (0.02 µCi/ml) for 24 hours, washed with nonradioactive medium and then incubated in fresh medium for at least 2 hours before drug treatment and the alkaline elution analysis for determination of the drug-induced ICL formation by the cisplatin-treated cells (13). The alkaline elution analysis enables evaluation of the relative lengths of the damaged DNA strands by determining the rate of elution of DNA from the filter. These independent experiments were replicated three times. The alkaline elution technique was performed as follows.
(1) Drug treatment
In the experiment on the formation of ICL by cisplatin, gastric cancer cells were incubated in 0, 5 and 20 µg/ml of cisplatin for 6 hours at 37℃ and then they were incubated in drug-free medium and the belotecan solutions at various concentrations.
(2) Alkaline elution assays
To introduce a known frequency of γ-ray-induced DNA single-strand breaks before analysis, approximately 3×105 drug-treated cells were irradiated with 6 Gy by 60Co γ-irradiation at a dose rate of 0.36 Gy/min. Before, during and after irradiation, the cells were kept on ice to prevent DNA repair. For the assay, the cells were diluted with cold DPBS (137 mM Na Cl:2.7 mM KCl:8.1 mM Na2HPO4:1.5 mM KH2PO4) and then deposited gently onto a polycarbonate filter (2.0- µm pore size, 25-mm diameter. Nucleopore Corp., Pleasanton, CA). The cells were lysed on the filter for 1 hour with 5 ml lysis solution that was comprised of 2% w.v SDS, 25 mM disodium EDTA, 50 mM Tris, 50 mM glycine and 0.5 mg/ml proteinase K (pH 9.6), which was allowed to flow through the filter by gravity, and then the filter was rinsed three times with 3 ml 20 mM disodium EDTA (pH 9.6) to remove most of the cell protein, membranes and RNA. The remaining DNA (over 97% of that applied to the filter) was eluted with tetrapropylammonium-tetrahydroxy-EDTA (pH 12.1) at a constant flow rate of 0.025~0.030 ml/min. Ten eluate fractions were collected directly into scintillation vials on a fraction collector at 1.5 hours intervals for 15 hours. Each fraction was mixed with 5 volumes of scintillation cocktail solution (Packard, Inc. USA) that contained 0.5% v/v acetic acid, and the radioactivity was counted using a LS3801 liquid scintillation counter. We examined the ICL formation induced by cisplatin with belotecan by performing alkaline elution assay at pH 12.1, which enabled the relative lengths of the damaged DNA strands to be evaluated by determining the rate of elution of DNA from the filter.
For quantitation of the DNA, the ICLs, which are the amounts of [14C]DNA from the sample cells retained on the filter, were calculated as a percentage of the amount retained after elution for 15 hours: these values were called the "relative retention". The frequency of CDDP-induced ICLs was calculated using the following formula:
CI=[(1-Rradiation)/(1-RCDDP)]1/2-1
in which CI is the CI of the CDDP-treated cells and Rradiation is the relative retention of 4-Gy irradiation. All of the values are expressed as means±SDs and they were analyzed using the two-tailed t test. p values<0.05 were considered to be statistically significant.
7) Preparation of nuclear extracts
The crude nuclear extracts were prepared was performed as follows. In brief, the cells were collected by centrifugation, washed twice with cold NB and resuspended in 1 ml cold NB; 9 ml cold NB containing 0.35% v/v Triton X-100, and 1 mM phenylmethyl-sulfonyl fluoride were then added. The cell suspension was kept on ice for 10 minutes and next washed with Triton X-100-free cold NB; the nuclear protein was eluted for 1 hour at 4℃ with cold NB that contained 0.35 M NaCl. The supernatant was obtained by centrifugation at 18,000×g for 10 minutes. The protein concentration was determined using the Bradford method.
8) DNA topoisomerase I activity
The activity of DNA topo-I was determined by measuring the relaxation of supercoiled Escherichia coli DNA (pBR322), which was performed as follows (14). For measurement of the total topo-I activity in the SNU-16 and SNU-601 cells, the reactionmixture was comprised 100 mM KCl, 10 mM MgCl2, 1 mM dithithreitol, 0.1 mM EDTA, 10% v/v glycerol, and 50 Tris-HCl (pH 7.4), 0.7 µg pBR322, and the crude nuclear extract (2 µg/ml protein). The reaction mixture used for measuring the inhibition of DNA relaxation by the topoisomerase I inhibitors was made by the addition of the drug solution or the equivalent volume of water to the reaction mixture. The reaction mixtures were incubated at 37℃ for 10 minutes, and the reactions were terminated by adding 45 µl of dye solution that was comprised of 2.5% w/v SDS, 0.01% w/v bromphenol blue, and 50% v/v glycerol. The mixtures were applied to 0.7% w/v agarose gels and electrophoresed for 4 hours with a running buffer of Tris-acetate EDTA. The gel was stained with 2 µM ethidium bromide and then photographed under transillumination with 300 nm UV light.
RESULTS
1) The cytotoxic effects of belotecan and cisplatin
The cytotoxicity of belotecan and cisplatin was assayed in the SNU-5, SNU-16 and SNU-601 cells by performing MTT assay. The IC50 values of belotecan against the SNU-5, SNU-16 and SNU-601 cells were 0.09375 (±0.001) µg/ml, 0.1875 (±0.005) µg/ml and 0.1875 (±0.003) µg/ml, respectively. These results were similar with those of previously reported by Lee et al (7), which showed the significant cytotoxicity of belotecan in SNU gastric cancer cell lines. The IC50 values of cisplatin against SNU-5, SNU-16 and SNU-601 were 6.00 (±0.04) µg/ml, 6.00 (±0.036) µg/ml and 16.00 (±0.051) µg/ml, respectively.
2) The effect of the belotecan and cisplatin combination on cytotoxicity
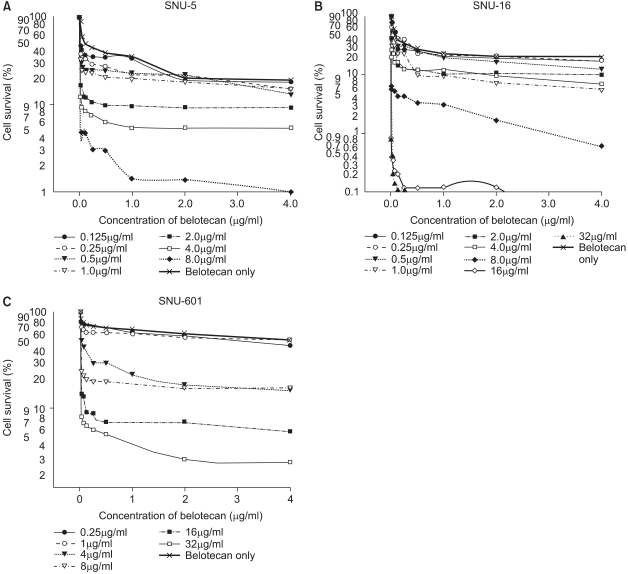
For the SNU-5 and SNU-16 cells, the cytotoxic effect of belotecan was much enhanced with the addition of an increasing dose of cisplatin. However, the cytotoxic effect of belotecan on SNU-601 cells was not markedly enhanced with the addition of cisplatin. The dose-response curves for the cytotoxicity by belotecan with different concentrations of cisplatin are shown in Fig. 3.
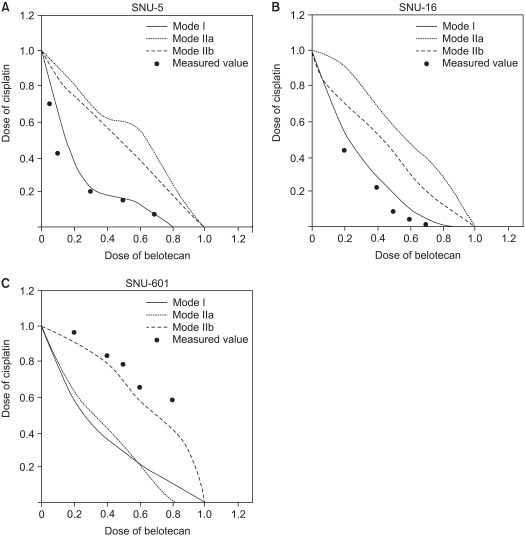
To determine whether the belotecan and cisplatin combination has synergistic cytotoxic effects, we drew the isobologramsat IC50 on the basis of each of the single-drug growth inhibitory curves and dose-response curves of belotecan (Fig. 4). The results showed that serial exposure to cisplatin and belotecan produced different effects on different cell lines. For the combinations of belotecan and cisplatin with SNU-16 cells, the data points fell in the area of supra-additivity. Most of the data points of the SNU-5 cells fell at the line of isobologram for the combination of belotecan and cisplatin. In case of the SNU-601 cells, the data points fell in the area of sub-additivity and even protectivity. These represent that there was synergistic interaction between belotecan and cisplatin for SNU-16 cells, but not for the SNU-5 cells or the SNU-601 cells.
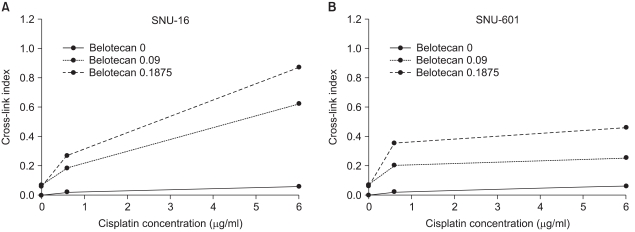
3) The role of belotecan on cisplatin-induced ICL formation
Because the definite differences in the effect of drug combinations (synergistic or antagonistic) were estimated between SNU-16 cells and SNU-601 cells, we analyzed the mechanisms of interaction by the combination of belotecan and cisplatin. The γ-irradiation induced DNA single-strand breaks and it reduced the amount of DNA retained on the filter in a dose-dependent manner, whereas the treatment with cisplatin before irradiation increased this in a concentration-dependent manner, suggesting that cisplatin induced ICL formation and it caused no single-strand breaks. The CIs of the SNU-16 and SNU-601 cells, which were exposed to cisplatin with various concentrations of belotecan, are shown in Fig. 5. The CIs of the SNU-16 cells treated with cisplatin and belotecan were higher than the CIs of the cells treated with cisplatin alone (p<0.005). Yet the CIs of the SNU-601 cells treated with both drugs were not significantly different from the CIs of the cells treated with cisplatin alone. These results suggest the hypothesis that the addition of belotecan can contribute to enhancing the formation of CIs by cisplatin in SNU-16 cells.
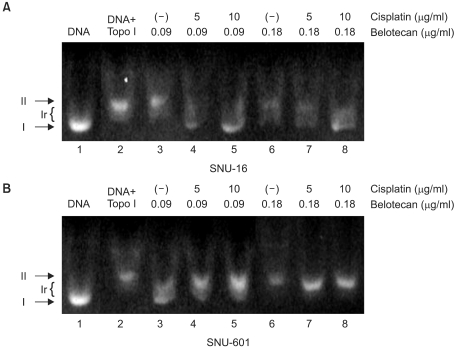
4) The role of cisplatin on the inhibitory effects of belotecan on topo-I
We also evaluated the role of cisplatin on the inhibition of topo-I by belotecan. The results are shown in Fig. 6. Fig. 6 represents the inhibitory effects of belotecan with and without cisplatin on the catalytic activity of topo-I. Over 90% of the plasmid DNA was in the superhelical form (Fig. 6A, Lane 1; Fig. 6B, Lane 1) in the condition that there was no addition of nuclear extract or drugs. The nuclear extract (2 µg/ml), which is where the topo-I was, from the SNU-16 and SNU-601 cells relaxed the supercoiled DNA (Fig. 6A, Lane 2; Fig. 6B, Lane 2); this was already expected. Yet the addition of belotecan inhibited the relaxation of the supercoiled DNA in the SNU-16 and SNU-601 cell lines (Fig. 6A, Lanes 3 and 6; Fig. 6B, Lanes 3 and 6), because of the inhibitory function of belotecan. Consequently, the addition of cisplatin (5 and 10 µg/ml) increased the topo-I inhibitory effect of belotecan in the SNU-16 cells (Fig. 6A, Lanes 4, 5, 7 and 8), but this showed no effects on the inhibition of topo-I by belotecan in the SNU-601 cells (Fig. 6B, Lanes 4, 5, 7 and 8). These results can be interpreted as that the difference of the effect of cisplatin between the SNU-16 and SNU-601 cells is correlated with the difference of the interaction between belotecan and cisplatin.
DISCUSSION
In this study, we demonstrated the synergistic interaction between belotecan and cisplatin against several gastric cancer cell lines. This study was carried out as a pre-clinical screening of potentially valuable belotecan combinations with cisplatin against gastric cancer. The combination of cisplatin and topo-I inhibitor seems to be an interesting strategy for cancer chemotherapy. The effect of this combination on various types of cancer has been reported in many previous studies (4,7,10,11,15). The concentration-response curves of anticancer agents with their nonlinear dose-response relationships do not always follow first-order kinetics. The additivity range of these drugs cannot be visualized by a simple additive method. Therefore, we used the isobologram method to investigate the effects of a combination of belotecan and cisplatin (12).
We found that belotecan had a supra-additive (synergistic) effect on SNU-16 cells, but it had a sub-additive (antagonistic) effect on SNU-601 cells when combined with cisplatin. The results for SNU-5 cells can be considered that belotecan has an additive effect rather than a synergistic effect because most of measured values by combined exposure were located at the isoeffect line. We chose the SNU-16 and SNU-601 cell lines for evaluating the mechanisms of interactions because of the definite difference between the results of the isobologram of the two cell lines.
As previously mentioned, we tried to elucidate not only the combined effect of belotecan ("new topo-I inhibitor") and cisplatin, but also the molecular mechanism responsible for this effect. The anticancer activity of cisplatin is caused by the formation of DNA adducts, which include DNA protein cross-links, DNA mono-adducts and interstrand and intrastrand DNA cross-links (16). To analyze the effect of belotecan on the cytotoxicity of cisplatin against gastric cancer cells, we used the alkaline elution test (13). If the value of the CIs is increased with the addition of drug A to cisplatin, the drug A is considered to have a positive effect on the formation of ICLs by cisplatin (13). In the present study, the CIs of the SNU-16 cells treated with cisplatin and belotecan were higher than the CIs of the SNU-16 cells treated with cisplatin alone (p<0.005). Furthermore, the higher concentrations of belotecan induced a greater difference of CIs in SNU-16 cells. In the case of SNU-601 cells, no significant differences of CIs were observed with the combination of belotecan. This result represents the positive effect of belotecan on the ICL formation induced by cisplatin, which is believed to be one of the mechanisms of the supra-additive effect of belotecan with cisplatin for some gastric cancer cells (11).
Minoru et al (11) demonstrated that with NB-506 treatment, topo-I inhibitor increased the ICL formation induced by cisplatin. They introduced three possible reasons for the enhancing effect of topo-I inhibitor on ICL formation; a) an increased cisplatin: nucleotide ratio around the DNA, b) increased affinity of DNA for cisplatin, and c) decreased repair after cisplatin adduct formation (11). The repair of cisplatin adducts is primarily mediated by the nucleotide excision repair pathway (17), and some drugs have been reported to interfere with excision repair. Arabinofuranosylcytosine, hydroxyurea and 5-FU inhibit the repair of cisplatin-induced ICLs (18,19). Although the definite mechanisms of the positive effect of belotecan on cisplatin remains unclear, these results indicate that belotecan may also mainly interfere with the repair of cisplatin-induced ICLs. It is possible that belotecan may interfere with DNA repair proteins that remove the cisplatin-induced DNA adducts, thereby increasing the cross-linking of DNA with cisplatin and enhancing its antitumor activity, and especially in SNU-16 cells. In contrast, enhanced cross-link repair has been suggested as a possible mechanism of cisplatin resistance. Previous studies have suggested an inverse relationship between the sensitivity to cisplatin and the repair of cisplatin adducts (20,21). The mechanisms of the subadditivity in SNU-601 cells could be explained by the enhanced cross-link repair.
The effect of cisplatin on the topo-I inhibitory effect by belotecan was estimated by performing DNA topo-I activity testing. Cisplatin (5 and 10 µg/ml) increased the inhibitory effect of topo-I by belotecan in SNU-16 cells, but not in SNU-601 cells. Since the inhibition of topo-I-mediated DNA relaxation by CPT analogues correlates with their antitumor activities (22), the result of this study suggests that cisplatin might enhance the inhibition of topo-I catalytic activity by belotecan.
The mechanisms for the modulating effect by cisplatin on the inhibition of topo-I catalytic activity by belotecan has not been precisely defined. A previous study on X-ray diffraction of the cross-linked dinucleotide cis-Pt(NH3)2(d(pGpG)) revealed that the 2 guanines were completely destacked and the deoxyribose sugar of the 5'-deoxyguanosine was in a C3'-endo pucker (23). This finding suggests that the intrastrand cisplatin cross-link (cross-linked dinucleotide cis-Pt(NH3)2(d(pGpG))) produces a severe local distortion in the DNA double helix, which leads to unwinding and kinking. The intrastrand cisplatin cross-link may also modulate the stabilization of topo-I-drug-DNA cleavable complexes, which interferes with the anticancer effect of belotecan. Additionally, Robert et al (24) have demonstrated a possible mechanism about the synergistic effect of cisplatin with camptothecin. They provided direct evidence for Pt-1,3-d (GTG) poisoning of topo-I in vitro and they demonstrated that persistent Pt-DNA adducts correlate with increased covalent topo-I-DNA complexes in vivo. Their results could indicate that the cytotoxic activity of cisplatin is due, in part, to poisoning of topo-I, which is exacerbated in the presence of topotecan (24). We didn't show the result about the topo-I inhibiting effect of cisplatin without belotecan in this article, but we also confirmed the topo-I inhibition by only cisplatin in our previeous experiment. Therefore, we could consider the enhancing cytotoxic effect of belotecan by cisplatin as "synergism" rather than "potentiation".
We didn't investigate why the combination effect is different among the different cell lines, and especially in the SNU-601 cells. The possible suggestion is the origin of the SNU-601 cells. The origin of SNU-601 cells was a signet ring cell carcinoma cell of a patient's ascites, who was previously treated with chemotherapy (unknown regimen) (25). Although the MDR gene has not been demonstrated in the SNU-601 cell line, this resistance is supposed to be acquired with previous chemotherapy.
The present study established the synergistic interaction between belotecan and cisplatin in gastric cancer. Although in vivo preclinical and clinical studies need to be conducted, our results have demonstrated the usefulness of this combination regimen (belotecan and cisplatin) for treating gastric cancer. Therefore, we consider the combination of belotecan and cisplatin as one of the options for the treatment of gastric cancer.
CONCLUSIONS
The combination of cisplatin with belotecan showed therapeutic synergy against the SNU-5 and SNU-16, human gastric cancer cell lines. The biochemical mechanism responsible for these interactions is suggested to be: a) belotecan modulates the repair of cisplatin-induced DNA adducts and possibly that b) cisplatin enhances the topoisomerase I inhibitory effects of belotecan. The combination regimen of belotecan and cisplatin can be considered as a chemotherapeutic treatment for gastric cancer.