Introduction
Triple-negative breast cancer (TNBC) is a breast cancer subtype that does not express estrogen receptor, progesterone receptor, or human epidermal growth factor receptor 2; its characteristics are earlier age of onset, greater metastatic potential, is highly malignant, poor prognosis, and a high mortality rate [1,2]. At present, TNBC treatment mainly involves chemotherapy, and a considerable number of patients experience toxic adverse effects; moreover, given blind chemotherapy, drug resistance is easily developed and the recurrence rate is high. Therefore, the search for a new treatment target in TNBC is presently one of the most important clinical requirements.
Receptortyrosine kinase (RTK) including MET (MET protooncogene, receptor tyrosine kinase), AXL, epidermal growth factor receptor and so on is a high affinity cell surface receptor for many polypeptide, growth factors, cytokines, and hormones that plays a key role in the development and progression of many types of cancer [3]. The MET (MET protooncogene, RTK) proto-oncogene family is found on the long arm of human chromosome 7, and the protein product is an RTK [4]. The family has two members: RON (macrophagestimulating 1 receptor) and MET. MET plays an important role in cell proliferation, differentiation, carcinogenesis, and invasion and migration [5]. RON is a specific cell membrane receptor for human macrophage-stimulating protein (MSP) [6]. MET is a receptor for hepatocyte growth factor (HGF) [7].
RON has been implicated as playing an important role in the development of various tumors, including pancreatic cancer, colorectal cancer, non-small cell lung cancer, and prostate cancer [6]. Abnormal expression of RON has also been detected in clinical samples of primary breast cancer [8,9]. Subsequent molecular mechanism experiments have shown that abnormal RON expression leads to hyperphosphorylation of RON kinase, which activates various intracellular signaling cascades downstream [8,10,11]. These lead to increased cell proliferation, migration, invasion, and drug resistance. In addition, abnormal RON expression is a therapeutic target in breast cancer that many small-molecule inhibitors of RON are undergoing preclinical and clinical evaluation [8,12-14]. The preliminary results indicate that inhibiting RON signaling has the potential for treating TNBC. At present, there is a lack of research on the role of RON in TNBC, and the mechanism remains unknown.
Similarly, aberrant activation of MET is frequently found in tumors, including mutations, gene amplification, increased transcription, and enhanced ligand activation [7]. Some studies have found frequent MET overexpression in invasive breast cancer [15,16]. These studies suggest that MET activation and overexpression can be an independent risk factor in breast cancer. Based on related research on RON and MET in primary breast cancer, we are committed to exploring whether RON and MET also play important roles in TNBC [17,18].
Currently, small-molecule tyrosine kinase inhibitors (TKIs) against the MET proto-oncogene family are widely used. Due to the existence of a signal cross-talk mechanism between RON and MET, most TKIs can inhibit both RON and MET, and a small proportion can only inhibit MET alone, while almost no TKIs inhibit RON alone [6,19]. Among the TKIs, BMS-777607 is a MET-related inhibitor that acts on c-Met, Axl, Ron, and Tyro3 [20,21]. And it has a stronger inhibitory effect on RON than on MET; its half maximal inhibitory concentration (IC50) in cell-free testing is 1.8 nM (RON) and 3.9 nM (MET) [20]. INCB28060 is a novel adenosine triphosphate (ATP)-competitive MET inhibitor with a cell-free assay IC50 of 0.13 nM and no activity against RON [22]. Tivantinib (ARQ 197) is the first non-ATP–competitive MET inhibitor with a cell-free assay inhibitory constant (Ki) of 0.355 μM with little activity against RON [23].
The present study was designed to determine RON and MET expression in primary TNBC samples and to evaluate their potential as a predictive prognostic biomarker of TNBC. Our rationale is that understanding RON and MET expression in TNBC can aid dissection of the molecular mechanisms underlying TNBC malignancy and identify a new treatment strategy for TNBC.
Materials and Methods
1. Cell lines and reagents
The TNBC cell lines HCC1806, HCC2185, MDA-MB-231, and SUM52PE were from American Type Cell Culture (ATCC, Manassas, VA). All cell lines were authenticated in 2015 with cytogenesis analysis. The cell lines were cultured and supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37°C. Mouse antiRON mAb (Zt/g4 and Zt/f2) and rabbit anti-RON (5029) antibody were used as previously described [8,24]. Human mature MSP and HGF were from R&D Systems (Minneapolis, MN). Phosphorylated (phospho)-tyrosine mouse mAb, phospho-MET, AKT, phospho-AKT, extracellular signal–regulated kinase (ERK) 1/2, and phospho-ERK1/2 were from Cell Signaling Technology (CST, Danvers, MA). All TKIs were from MedchemExpress (MCE, Monmouth Junction, NJ). BMS-677007, INCB28060, and tivantinib were dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 10 mM and stored at –20°C.
2. Patients and tissue specimens
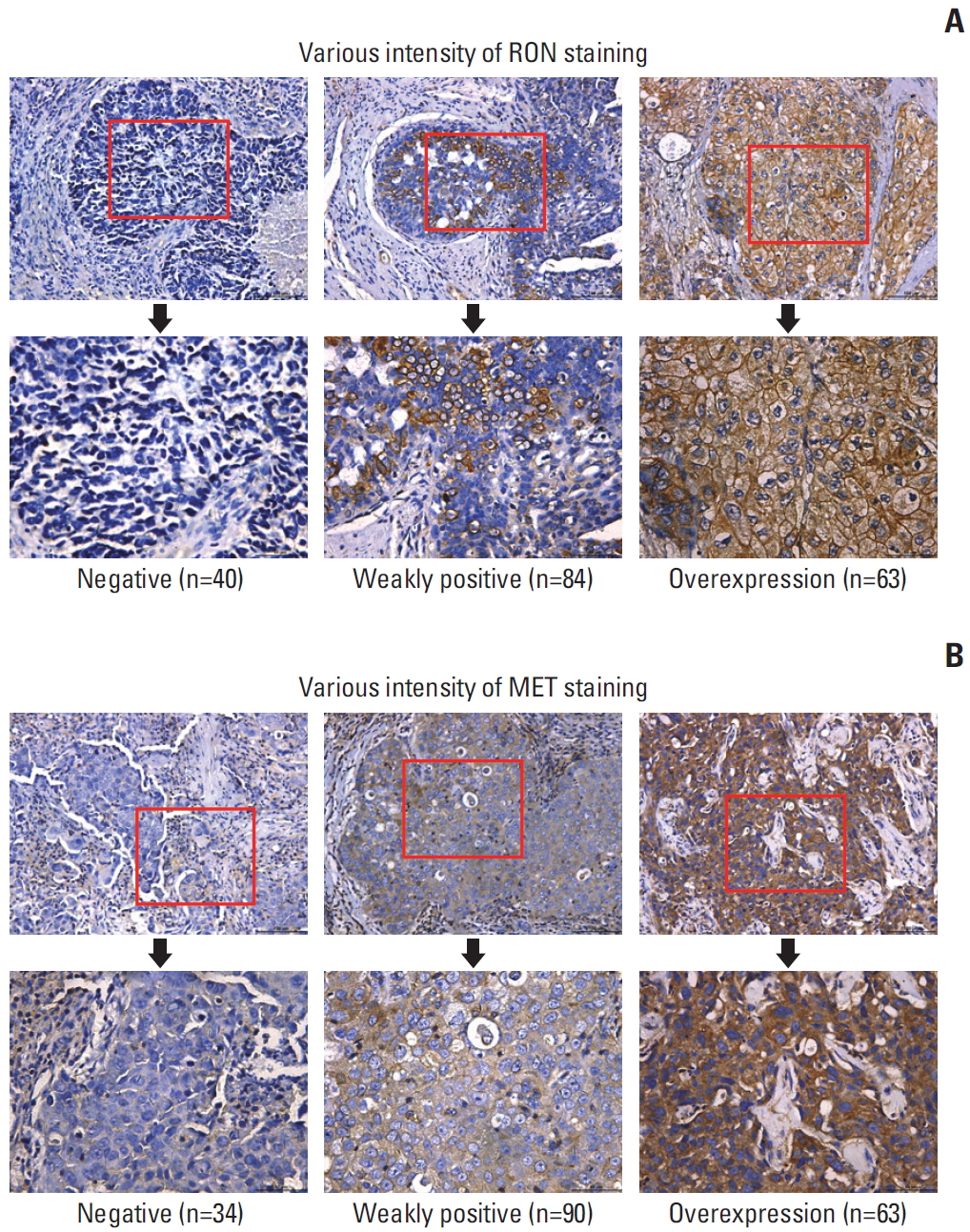
We analyzed 187 patients pathologically diagnosed with TNBC between May 2011 and October 2015 at The First Affiliated Hospital, Zhejiang University School of Medicine. All patients underwent pathological biopsy for breast cancer surgery. The clinical parameters included patient demographics, tumor-node-metastasis (TNM) stage, tumor differentiation, tumor size, and treatment modality. All tissues were fixed in 10% buffered formalin and embedded in paraffin. Immunohistochemical (IHC) staining was carried out using Zt/f2 for RON as described previously and using rabbit anti-MET mAb for MET, followed by reagents from Envision System (Dako, Carpentaria, CA) [24]. Two pathologists reviewed all archival hematoxylin and eosin–stained sections. This method combines scores from staining intensity (0-3) and the proportion of stained cells (0-4). A combined score of 2-4 was deemed weakly positive, while a combined score ≥ 5.0 was considered high expression or overexpression. Patients with RON overexpression and MET overexpression were considered to have co-overexpression.
3. Flow cytometry
Individual TNBC cell lines were digested before immunofluorescence staining antibody treatment. Surface RON was detected using Zt/g4 after combination with anti-mouse IgG conjugated with Alexa Fluor 488 (BD, New York, NY); MET was detected using anti-MET mAb conjugated with BV510 (BD). Normal mouse IgG was used as the isotype control. All flow cytometry experiments were performed using BD FACSCanto II (BD).
4. Cell viability and cellular caspase-3/7 activity assays for apoptosis
HCC1806, HCC2185, SUM52PE, and MDA-MB-231 cells (8,000 cells per well in 96-well plates in triplicate) were treated with different concentrations of BMS-777607, INCB28060, or tivantinib for 96 hours. Cell viability after TKI treatment was determined using Cell Counting Kit-8 (CCK-8; MCE). DMSO-treated cells were used as the control. Caspase-3 and caspase-7 activity was measured using the caspase-Glo 3/7 assay system (Promega, Madison, WI) according to the manufacturer’s instructions. The results are expressed as the fold induction of the control (caspase-3/7 activity of the control cells was set as 1).
5. Phosphorylation and western blotting
MDA-MB-231 cells were treated with the IC50 of BMS-777607, INCB28060, or tivantinib in serum-free medium. The phosphorylation assay was performed by stimulating the cells (2×106 cells/mL/sample) with 2 nM MSP and HGF at 37°C for 15 minutes. The cells were lysed in radioimmunoprecipitation (RIPA) buffer and separated by 8% sodium dodecyl sulfate–polyacrylamide electrophoresis (20 μg per sample) under reduced conditions. RON, MET, downstream pathway proteins including AKT and ERK1/2, and the phosphorylated proteins were detected using rabbit mAb as described above and visualized using enhanced chemiluminescent reagents. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control to ensure equal sample loading.
6. Cell motility assay
Cell motility was analyzed by treating MDA-MB-231 cells (2×105 cells per well in 6-well plates) with TKIs for 24 hours. The cells were serum-starved overnight and scratched to generate artificial gaps. The cells were treated with the IC50 of BMS-777607, INCB28060, or tivantinib in serum-free medium. Cell motility was monitored, and cells that had migrated into the denuded area were scored. Images were collected at 0, 6, 12, and 24 hours under an inverted microscope (Zeiss, Jena, Germany).
7. TNBC xenograft mouse model and TKI treatment
Female athymic nude mice (6 weeks old) were purchased from Shanghai Laboratory Animal Center, injected with 1×107 TNBC cells in the right subcutaneous space, and randomly divided into different groups (n=4 per group). Treatment was initiated when the average tumor volume was 100-150 mm3. Tumor-bearing mice were dosed orally by gavage daily with 25 mg/kg BMS-777607, 10 mg/kg INCB28060, or 20 mg/kg tivantinib for up to 15 days. Tumor volume (V) was measured every 3 days and calculated as follows: V=(length×width2)/2. Tumor growth was monitored for 51 days. Animals were euthanized when tumor volumes were > 2,000 mm3 or if the tumors became necrotic or ulcerated through the skin.
8. Statistical analysis
GraphPad Prism 7 (GraphPad Software, San Diego, CA) and SPSS ver. 17.0 (SPSS Inc., Chicago, IL) were used for statistical analysis. The relationships between RON and MET expression and the clinicopathologic characteristics were compared using the chi-square test, one-way analysis of variance (ANOVA), or the two independent samples t test. Overall survival (OS) was calculated as the time from diagnosis of TNBC until death or the date of the last follow-up. Survival data were analyzed by the Kaplan-Meier method and log-rank test. Results are reported as the mean±SD. The control and treatment group data were compared using Student’s t test. Statistical differences at p < 0.05 were considered significant.
Results
1. Clinical characteristics of patients
The clinical features of the 187 TNBC cases are as follows (Table 1): the patients were aged 27-83 years; the mean age was 52.82±12.44 years. The TNM stage distribution was: stage I, 85 cases (45.5%); stage II, 85 cases (45.5%); stage III, 17 cases (9.1%). Most patients (n=147, 78.6%) had poor pathological grading. Among the remaining patients, one case (0.5%) was well-differentiated, and 20 cases (10.7%) were moderately differentiated. The differentiation of the remaining 19 cases (10.2%) was unknown. One hundred and sixty-one patients (86.1%) received chemotherapy while the others did not. Forty-eight patients (25.7%) had lymph node metastasis during follow-up. Distance metastasis was confirmed in 19 patients (10.2%) during follow up.
2. RON and MET expression in TNBC
RON and MET staining were detected in the cell membrane and cytoplasm of cancer cells, while that of paracancerous cells were weakly stained. RON and MET immunoreactivity, shown by the different staining intensities, is shown in Fig. 1. One hundred and forty-seven cases (78.6%) were RON-positive; 63 cases (33.7%) had RON overexpression. One hundred and fifty-three cases (81.8%) and 63 cases (33.7%) were MET-positive and MET-overexpressing, respectively. One hundred and twenty-seven cases (67.9%) and 43 cases (23.0%) were RON/MET co-positive and RON/MET co-overexpressing, respectively. IHC scoring showed that RON and MET expression were correlated (Pearson correlation analysis, p < 0.01) (S1 Table). Table 1 summarizes the information on RON and MET expression in the TNBC samples. However, IHC staining confirmed that RON and MET were widely expressed in patients of different ages, TNM stage, pathological grade, and primary TNBC with or without distant or lymph node metastasis.
3. Relationship between RON and MET expression and OS
The role of RON and MET expression in OS was assessed using Kaplan-Meier analysis and the log-rank test (Fig. 2). We analyzed a total of 176 patients after excluding patients with other tumors or systemic diseases (such as cirrhosis, uncontrollable diabetes or hypertension, etc.). All patients were followed until May 2018. The 5-year survival rate of all patients was 83.9±3.1% while the 3-year survival rate of all patients was 88.4±2.4%. RON and MET expression levels were significantly correlated with patients' OS (p < 0.05 and p < 0.01 correspondingly). Patients with RON high-expression tumors had significantly worse 5-year survival rate (73.4%±6.3%) than patients with RON low-expression tumors (89.2%±3.2%) (Fig. 2A). Similarly, there was poorer survival among patients with MET high-expression tumors compared with patients with MET low-expression tumors (5-year survival rate, 76.1%±5.8% vs. 87.6%±3.5%) (Fig. 2B). To assess the relative importance of RON and MET co-expression, we combined the two biomarkers to evaluate their association with survival (Fig. 2C and D) and found that patients with RON+/MET+ tumors had worse survival compared with patients with RON–/MET–tumors (5-year survival rate: 73.5%±7.7% vs. 90.0%±3.5%, p < 0.01). The 5-year survival rate in patients with RON+/MET–tumors and RON–/MET+ tumors was 73.3%±10.6% and 80.7%±8.7%, respectively.
4. RON and MET expression in TNBC cell lines
The significance of RON and MET in the TNBC samples prompted us to determine if breast cancer cell lines also express RON and MET (S2 Fig.). Among the four TNBC cell lines, RON was expressed in SUM52PE, HCC2185, and MDA-MB-231 cells, while MET was expressed only in MDA-MB-231 cells.
5. TKIs inhibited TNBC cell proliferation modestly and induced apoptosis
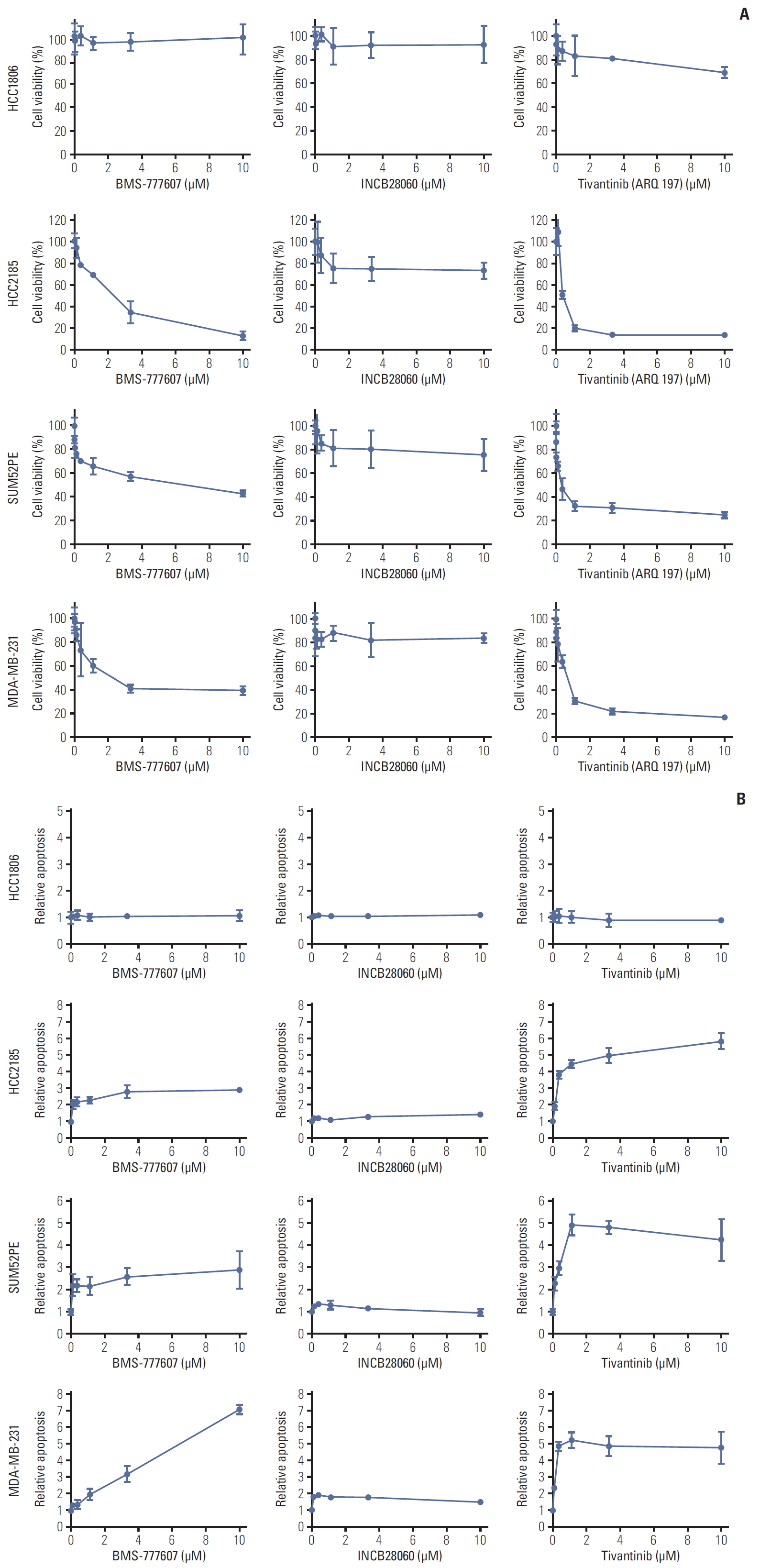
The TNBC cell lines were incubated with BMS-777607, INCB28060, or tivantinib for 96 hours. Fig. 3A shows the effect of the TKIs on TNBC cell viability. BMS-777607 and tivantinib reduced HCC2185, SUM52PE, and MDA-MB-231 cell viability significantly and in a dose-dependent manner. The IC50 of BMS-777607 were 1.925 μM, 3.727 μM, and 2.273 μM for HCC2185, SUM52PE, and MDA-MB-231, respectively. The IC50 of tivantinib were 0.5339 μM, 0.3656 μM, and 0.7293 μM for HCC2185, SUM52PE, and MDA-MB-231, respectively. Compared with the other two TKIs, INCB28060 had a weaker inhibitory effect, and its IC50 was > 10 μM for the HCC2185, SUM52PE, and MDA-MB-231 cells that was considered of no statistical significance. At 0-10 μM, all three TKIs had no significant effect on the growth of HCC1806 cells, which do not express RON or MET.
We also tested the effect of TKIs on TNBC cell apoptosis. Fig. 3B shows the apoptosis activity in the cell lines. BMS-777607 and tivantinib increased HCC2185, SUM52PE, and MDA-MB-231 cell apoptosis significantly in a dose-dependent manner. The median effect concentration (EC50) of BMS-777607 were 7.816 μM, 7.778 μM, and 2.766 μM for HCC2185, SUM52PE, and MDA-MB-231, respectively. The EC50 tivantinib were 0.2427 μM, 1.079 μM, and 0.3446 μM for HCC2185, SUM52PE, and MDA-MB-231 cells, respectively, while there was no significant difference in apoptosis between the 0-10 μM INCB28060 groups.
6. TKIs suppressed cell migration
All three TKIs significantly suppressed cell motility at their respective IC50 from 0 hour until 24 hours (Fig. 4). At this dose, the 24-hour mobility of cells treated with BMS-777607, INCB28060, and tivantinib was 44.15% (p < 0.001), 62.43% (p < 0.001), and 21.91% (p < 0.001), respectively, compared with the 95.99% in the control group. Compared with BMS-777607 and tivantinib, INCB28060 caused less inhibition of MDA-MB-231 cell migration (p < 0.001).
7. TKIs inhibited the RON- and MET-induced signaling pathways
MDA-MB-231 cells were treated with the TKIs at their respective IC50 for 30 minutes or 60 minutes and evaluated by western blotting (Fig. 5). BMS-777607 inhibited RON and MET phosphorylation, and the phosphorylation signals of downstream AKT and ERK1/2 were almost completely inhibited, while their total protein was unchanged. In contrast, INCB28060 only inhibited MET phosphorylation, and its inhibition of AKT and ERK1/2 phosphorylation was weaker than that of BMS-777607. On the other hand, tivantinib inhibited the basal levels of phosphorylated RON and MET within 30 minutes. Compared with BMS-677077, it did not obviously inhibit AKT and ERK1/2.
8. Therapeutic activity of TKIs in TNBC xenograft model
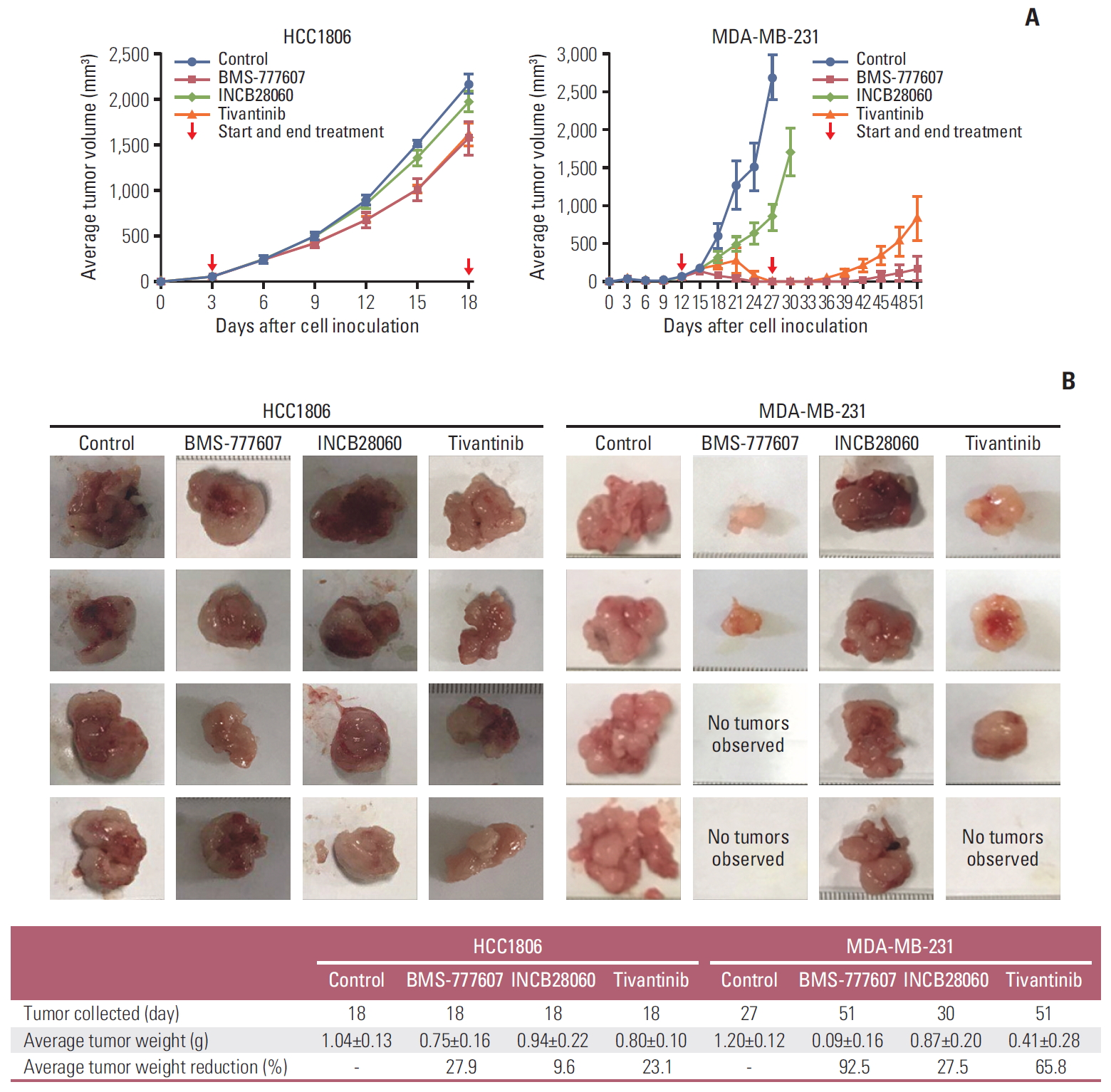
Of the four TNBC cell lines tested, HCC1806 and MDA-MB-231 cells could grow in athymic nude mice and were selected for the therapy studies. In our mouse tumor-bearing model, HCC2185 and SUM52PE cells did not successfully form tumors, which is both consistent with some studies [25] and inconsistent with others [26]. The HCC1806 cell–initiated xenografts grew rapidly and were insensitive to all three TKIs (Fig. 6A). The control and experimental groups had a tumor volume of 2,000 cm3 at around day 15 of dosing. After the mice had been sacrificed by cervical dislocation, the tumor tissues were removed; the tumor mass between the groups was not statistically significantly different. For MDA-MB-231 cells, INCB28060 could delay tumor volume to 2,000 cm3 (30 days vs. 27 days) while the tumor mass was not statistically significant (0.87±0.20 g vs. 1.20±0.12 g). BMS-777607 and tivantinib reduced the tumor significantly, and the average tumor volume was < 200 mm3 by 15 days. After treatment had been halted, the tumor growth rate of the BMS-777607 and tivantinib groups was slower than that of the control group. Fig. 6B shows the tumors obtained from the mice. Compared with the control group, the tumor growth reduction rate in the BMS-777607 group was 92.5% (p < 0.001); that in the tivantinib group was 65.8% (p < 0.001). Tumors in the tivantinib group grew faster than that in the BMS-777607 group (p < 0.001).
Discussion
The present study is our first attempt to analyze RON and MET expression in a large number TNBC samples. We show that RON and MET were widely overexpressed in TNBC samples regardless of patient age, pathological grade, clinical stage, metastasis, and tumor size. In our study, patients with RON overexpression tended to have higher Met expression, and vice versa, which encouraged us to study the cross-talk between RON and MET.
In the survival analysis, our studies suggest that RON and MET may be involved in an important process in the malignant progression of TNBC. Therefore, abnormal RON and MET could be used as biomarkers of poor prognosis in TNBC. Among the four groups, the 5-year survival rate of RON+/MET+ patients was significantly lower than that of RON+/MET–, RON–/MET+, and RON–/MET–. This prompted us to combine the two targets of RON and MET to comprehensively assess the prognosis of triple-negative breast cancer patients, not just in breast cancer [15]. With the gradual popularization of molecular diagnostics, we believe that detecting RON and MET expression in patients with TNBC in clinical practice would greatly important for predicting the malignant behavior of cancer.
Targeted RON TKIs require targets to have important physiological significance in tumor development [25]. As TKIs rarely inhibit RON alone, we selected BMS-777607 for the present study. Due to the much stronger inhibition of RON than MET, we believe that BMS-777607 can inhibit RON and MET, in which inhibition of RON plays a major role [20].
First, in the cell proliferation experiment, BMS-777607 significantly inhibited the proliferation efficiency of the RON-positive HCC2185, SUM52PE, and MDA-MB-231 cells with a dose-related correlation. The apoptosis experiments suggest that BMS-777607 inhibits cell proliferation by inducing apoptosis. The cell migration experiments confirmed that BMS-777607 can inhibit RON-mediated cell migration and invasiveness.
Second, BMS-777607 inhibited RON phosphorylation, and the phosphorylation signals of the downstream AKT and ERK1/2 were almost completely inhibited. In previous studies, RON activation stimulated two signaling pathways: RAS-ERK1/2 and phosphoinositide 3-kinase (PI3K)–AKT. The RAS–ERK pathway is essential for regulating RON-mediated activities such as growth, survival, and invasiveness to cause epithelial-to-mesenchymal transition [27-29]. The PI3K-AKT pathway regulates RON-mediated cellular invasive growth [30].
Third, we confirmed the therapeutic efficacy of BMS-777607 in a mouse TNBC xenograft model. It is quite likely that BMS-777607 inhibits tumor growth and kills tumors both in vitro and in vivo. Moreover,some studies suggested inhibition of Axl is also main mechanism for BMS-777607 and Axl is highly expressed in the TNBC tumor cell line MDAM-BA-231 [21,31]. These findings lay the foundation for BMS-777607 targeting Tyrosine kinase to treat TNBC into clinical trials in the future.
In the following study, we selected two small-molecule inhibitors, i.e., INCB28060 and tivantinib, which inhibit MET alone and do not inhibit RON [22,23], and our experiments yielded near opposite results.
Tivantinib inhibits cell proliferation and induces apoptosis, whereas INCB28060 does not. In the cell migration experiment, the tivantinib group had a significantly higher inhibition rate than the INCB28060 group. Second, unlike BMS-777607 targeting of RON, INCB28060 and tivantinib inhibited the downstream ERK1/2 and AKT weaklier. Related studies have shown that, similar to RON, the signaling pathways downstream of MET have both RAS-ERK1/2 and PI3K-AKT pathways [7,27]. Finally, tivantinib inhibited MDA-MB-231 xenograft growth in the nude mice, whereas INCB28060 did not. Tivantinib also targets MET, where it can inhibit TNBC in vitro and in vivo.
On the other hand, INCB28060 was barely effective. Due to the multi-locus nature of small-molecule drugs, we believe that the anti-tumor effect of tivantinib is not MET-dependent and may be related to other targets. A recent study reported that the potent anti-tumor activity of tivantinib in hepatocellular carcinoma was achieved by targeting microtubules [32].
RON and MET can cross-talk and cooperate in intracellular signaling via transphosphorylation by forming specific RON-MET complexes [33]. This is consistent with the fact that the TKIs specifically targeting MET in the present study can slightly inhibit RON phosphorylation at cellular level. These basic research studies have also laid the foundation for the joint inhibition of RON and MET in TNBC such as BMS-777607 and tivantinib.
In summary, RON and MET are widely expressed in TNBC cancer tissues. This study demonstrates that RON and MET may be involved in an important process in TNBC malignant progression and can be important biomarkers in TNBC of poor prognosis. TKIs targeting RON and Met can inhibit the activation of the downstream signaling molecules, inhibit TNBC cell migration and proliferation, and increase TNBC cell apoptosis in vitro; in nude mouse xenograft models, the TKIs can significantly inhibit tumor growth and shrink tumor volume. TKIs targeting RON and MET have stronger drug development potential in TNBC treatment.