Introduction
The study of the association between diet and human cancer has drawn considerable attention in recent decades, especially which and how dietary factors might influence cancer risk. The new concept that changing diet can alter the epigenetic state of genes, thereby increasing or decreasing cancer risk, has been put forth in recent years [1]. Epidemiological studies have indicated a connection between various fatty acids (FAs) and ovarian cancer risk [1]. It has been reported that polyunsaturated fatty acids (PUFAs) can generate several omega-3 lipid metabolites and inhibit omega-6 arachidonic acid production, thus playing a beneficial role [2]. Previous study has shown the benefit of the omega-3 FAs on breast cancer incidence [3], but little information exists on the effects and mechanisms of eicosapentaenoic acid (EPA) in ovarian cancer cell growth.
Ovarian cancer is the most fatal gynecologic malignancy worldwide [4]. It is mostly detected at an advanced stage, leading to a high mortality rate. The pathogenesis of ovarian cancer has long been investigated but is still poorly understood. Obviously, it is urgent to optimize or develop new therapeutic strategies in the management of this disease. EPA, one kind of omega-3 polyunsaturated FA, has been observed to be significantly lower in the plasma of patients with any of several cancers than in the plasma of healthy individuals [4], implying its considerable role in cancer development. Several studies have suggested that EPA inhibits ovarian cancer cell growth and suppresses tumor metastasis [5].
Compelling evidence shows that FAs could achieve their biological effect by governing the fluidity and configuration of membrane receptors, cell signaling pathways, transcription of genes, and inflammatory response [6]. In recent years, many studies have described and characterized several G protein–coupled receptors (GPRs) for FAs in cancer cells, which have been reported to be critical components of the sensing apparatus in the human body and to have specificities for FAs of differing saturation degrees and chain lengths. Free fatty acid receptor 1 (GPR40) and GPR120, which both have Gαq/11 as their Gα subunit, are receptors for long-chain unsaturated FAs. GPR30, another novel FA receptor, is a 7-transmembrane estrogen receptor of the Gαs family predominantly localized in the endoplasmic reticulum. Many steroids and some synthetic estrogen-receptor ligands function by binding to GPR30 [7]. Once bound, the production of cyclic AMP (cAMP) was promoted, causing epidermal growth factor cleavage and epidermal growth factor receptor transactivation [7]. GPR30 is expressed not only in normal tissues, including the uterus, ovaries, and mammary glands, but also in several cancers, including ovarian cancer, in which GPR30 is highly expressed and correlated with a poor prognosis [8].
In this study, we found that EPA suppressed ES2 ovarian clear cell carcinoma (OCCC) cells growth through GPR30 by activating caspase-3 and inducing apoptosis. Furthermore, GPR30 was expressed in patients with various ovarian cancers, and 300 μM EPA suppressed human ovarian cancer cell growth via GPR30 without influencing normal cells. All these findings open a new front in the battle against ovarian cancer.
Materials and Methods
1. Materials
FAs, FA-free bovine serum albumin (BSA), and forskolin were purchased from Sigma-Aldrich (St. Louis, MO). Fura2/AM was purchased from Dojindo (Kumamoto, Japan). G1 and G15 were purchased from TOCRIS Bioscience (Ellisville, MI) and Merck (San Diego, CA), respectively. YM254890 was from Yama-nouchi Pharmaceutical Co., Ltd. (Ibaraki, Japan).
2. Cell lines and culture
ES2 and SKOV3 cell lines were obtained from the American Type Culture Collection (ATCC). ES2 and SKOV3 cells were cultured in McCoy’s 5A medium supplemented with 10% fetal bovine serum and antibiotics. Cells were cultured in Opti-MEM (Gibco, Grand Island, NY) without fetal bovine serum. FAs were first dissolved in pure ethanol and then freshly diluted to the culture medium containing 1% BSA. The ethanol and BSA were 0.05% (v/v) and 0.1% (v/v), respectively.
3. Cell proliferation assay and cytotoxicity assay
For the assay of ES2 and SKOV3 cell proliferation, 5×103 cells per well in Opti-MEM with different types and concentrations of FAs were seeded in triplicate, and cell numbers were monitored after 24, 48, and 72 hours using the Cell Counting Kit-8 (Dojindo). The cytotoxicity of various FAs at different concentrations on ES2 and SKOV3 cells was measured with the Cytotoxicity LDH Assay Kit-WST (Dojindo). All experiments were performed in triplicate wells for each condition and repeated three times independently.
4. Cell apoptosis assay
A total of 0.5×106 cells were cultured into 6-well culture plates and cocultured with or without 300 μM EPA for 48 hours. For cell apoptosis analysis, cells were determined with the Annexin V/PI Apoptosis Detection Kit (Dojindo) and analyzed by a FACSCalibur (BD Bioscience, San Jose, CA).
5. Cytosolic calcium determination
Cells were trypsinized in 3.5-cm Petri dishes, centrifuged, and resuspended in phosphate buffered saline (PBS) with 3 mM Fura-2/AM. Cells were treated with the Ca2+ indicator Fura-2/AM (5 μM) to measure [Ca2+]i. After washed for three times to remove extracellular Fura-2/AM, the Fura-2 was detected using Felix fluorescence data acquisition as previously shown [9]. The dose of YM254890 was based on previous findings [10,11].
6. cAMP measurement
Cells were seeded and treated with G15 or GPR30 small interfering RNA (siRNA). The dose of G15 was based on Wang et al. [12] and Bai et al. [13], who found that the 8 μM dose would be sufficient to obtain antagonistic effects on GPER1/GPR30 without affinity for other estrogen receptors. Twenty-four hours later, the cells were seeded in Opti-MEM medium for 5 hours and then treated with BSA, palmitic acid, or EPA. Cells were washed with PBS twice and frozen and thawed three times after treatment. The cAMPs were quantified with an Enzyme Immu-noassay Kit (Cayman Chemical, Ann Arbor, MI).
7. Lipid-protein overlay assay
The nitrocellulose membrane was spotted with EPA (1, 3, and 5 mM) and then blocked in buffer as previously reported [14]. Next, the membrane was incubated with purified GFP-GPR30 protein (100, 200, and 400 ng/mL) overnight at 4°C, then scanned by Typhoon FLA9500 (GE Healthcare, Uppsala, Sweden) to detect GFP.
8. Ovarian cancer xenograft models
Ovarian cancer xenograft models were generated in old female nude mice at 4 to 6 weeks of age by injecting 1×107 ES2 cells treated with or without adenoviral GPR30 shRNA into subcutaneous fat. Animals were divided into four groups with 10 per group. For the experimental group, 10 μL EPA was added to 90 μL aqueous vehicle. For the control group, 10 μL ethanol was added to 90 μL aqueous vehicle. Mice were given EPA (4 μg) once daily. For the treatment of G15, ovarian cancer xenograft models were generated in old female nude mice at 4 to 6 weeks of age by injecting 1×107 ES2 cells into subcutaneous fat. Animals were divided into four groups with 10 per group. Nude mice bearing ovarian tumors were received dimethyl sulfoxide (DMSO) in combination with MeOH as a control, EPA in combination with DMSO, MeOH in combination with G15 or EPA in combination with G15. Mice were given EPA (4 μg) once daily, while G15 (10 μg) once daily from day 5. Tumor volumes were calculated as 1/2×length×width2. Ki67 and GPR30 were stained to assay the inhibitory effects of EPA and GPR30 expression, respectively.
9. Tissue microarray construction and immunohistochemical analysis
Ovarian cancer patient (n=173) samples were collected from the Department of Pathology of Nanjing Drum Tower Hospital. The tissue microarray (TMA) was made as previously reported [15]. The immunohistochemical stains were assessed by three separate observers who did not know patient characteristics. The classification of GPR30 expression level was based on the staining intensity of GPR30. All protocols for humans were reviewed and approved (kyy2015-38). The detailed information of TMA was provided in S1-S3 Tables.
10. Quantitative real-time PCR
Quantitative real-time PCR was performed using SYBR Select Master Mix (Vazyme, Nanjing, China) in an ABI 7300 sequence detector. The results were normalized to the 18S rRNA gene expression level in each sample. The primers were from PrimerBank (http://pga.mgh.harvard.edu/primerbank/).
11. Antibodies and immunoblotting
The antibodies used were cleaved caspase-3, AKT, p-AKT, ERK, p-ERK, phosphorylated protein kinase A (p-PKA) substrate (Cell Signaling Technology, Danvers, MA), actin (Sigma-Aldrich), and GPR30 (Santa Cruz Biotechnology, Santa Cruz, CA).
12. RNA interference
The siRNA against GPR30 was GCACCUGUGGCUGACGAAUUU. ES2 cells were transfected with RNA interference (RNAi) oligonucleotides using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA).
13. Adenoviral expression vectors
The GPR30 shRNA sequence was GCACCTGTGGCTGACGAATTT. Adenovirus was amplified and purified using Sartorius Adenovirus Purification kits (Sartorius, Germany).
14. Statistics
Data from at least three independent experiments were analyzed and are expressed as means±standard deviation. The nonpaired Student’s t test was used for these analyses. A difference was considered significant at p < 0.05, p < 0.01, and p < 0.001. R was used for the correlation analysis of GPR30 expression level with overall survival of ovarian cancers patients. Then packages “survival” and “survminer” were used for survival analysis using Kaplan-Meier method and generate the survival curve.
Results
1. EPA inhibits cell proliferation in ovarian cancer cells
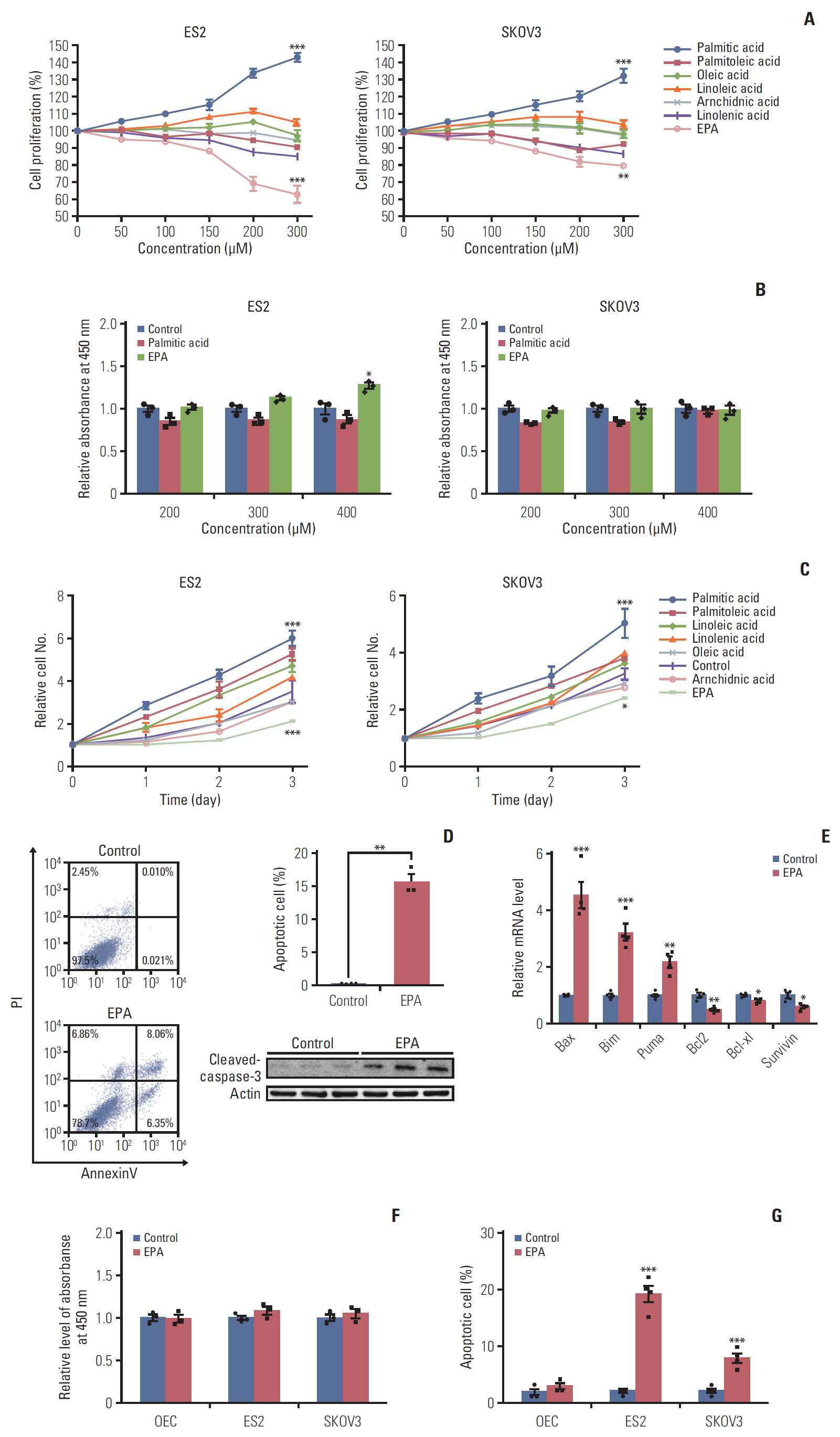
FAs have an effect on cancer by altering cell membrane synthesis and saturation and via cholesterol lipid hormones that affect the function and distribution of signaling macromolecules [6]. Thus, we speculated that various FA treatments might influence different aspects of cancer development in ovarian cancer cells. Ovarian carcinoma is divided into four major histological types: clear cell, mucinous, serous, and endometrioid [16]. OCCCs makes up 25% of ovarian carcinoma in Japan, and the lethality rate of adenocarcinoma is also high [16]. Compared with other types of ovarian carcinomas, OCCCs have a poorer prognosis and are more frequently platinum resistant [17]. Here, we selected the ES2 OCCC and SKOV3 adenocarcinoma cell lines for study. ES2 and SKOV3 were first treated with several FAs at different doses for 24 hours (0-300 μM). We found that palmitic acid, a saturated FA, obviously enhanced cell growth, while EPA inhibited ES2 and SKOV3 cell proliferation in a dose-dependent manner (Fig. 1A), which is in accordance with the published anticancer effects of EPA [18]. However, EPA promoted cell proliferation in CAOV3 and A2780 cell lines, indicating the antitumor effect of EPA may be cell line specific event (S4 Fig.). We also analyzed the toxicity of palmitic acid and EPA on ES2 and SKOV3 cells by using a cytotoxicity assay. We found that cell toxicity affected ES2 cells at a 400 μM concentration, while the effect on SKOV3 cells was minor at 400 μM (Fig. 1B). Thus, 300 μM was an appropriate concentration for use in subsequent experiments. The pro-proliferation effect of palmitic acid and the inhibitory effect of EPA on ES2 and SKOV3 cells were also time-dependent. Palmitic acid treatment increased cell growth in both ovarian cancer cell lines. However, the growth of ES2 cells was significantly decreased when treated with EPA compared to that of SKOV3 cells (Fig. 1C), suggesting that EPA had a much more pronounced effect in ES2 cells than in SKOV3 cells.
As previously reported, n-3 PUFAs (docosahexaenoic acid or EPA) inhibit colorectal cancer cell proliferation by inducing cell apoptosis [19]. Therefore, we detected cell apoptosis caused by EPA treatment in ES2 cells. The flow cytometry results showed that apoptotic cells were approximately 14.4% in EPA-treated ES2 cells, more than that in control (0.03%). The protein expression analysis also confirmed that EPA led to cleaved caspase-3 activation (Fig. 1D). mRNA expression analysis confirmed that EPA-induced proapoptotic genes activation and antiapoptotic genes inhibition in ES2 cells (Fig. 1E). Importantly, the cytotoxicity and apoptosis of normal ovarian epithelial cells were not affected by 300 μM EPA treatment (Fig. 1F and G). Together, these data indicate that EPA induces ovarian cancer cell apoptosis.
2. GPR30 acts as a novel EPA receptor in ES2 cells
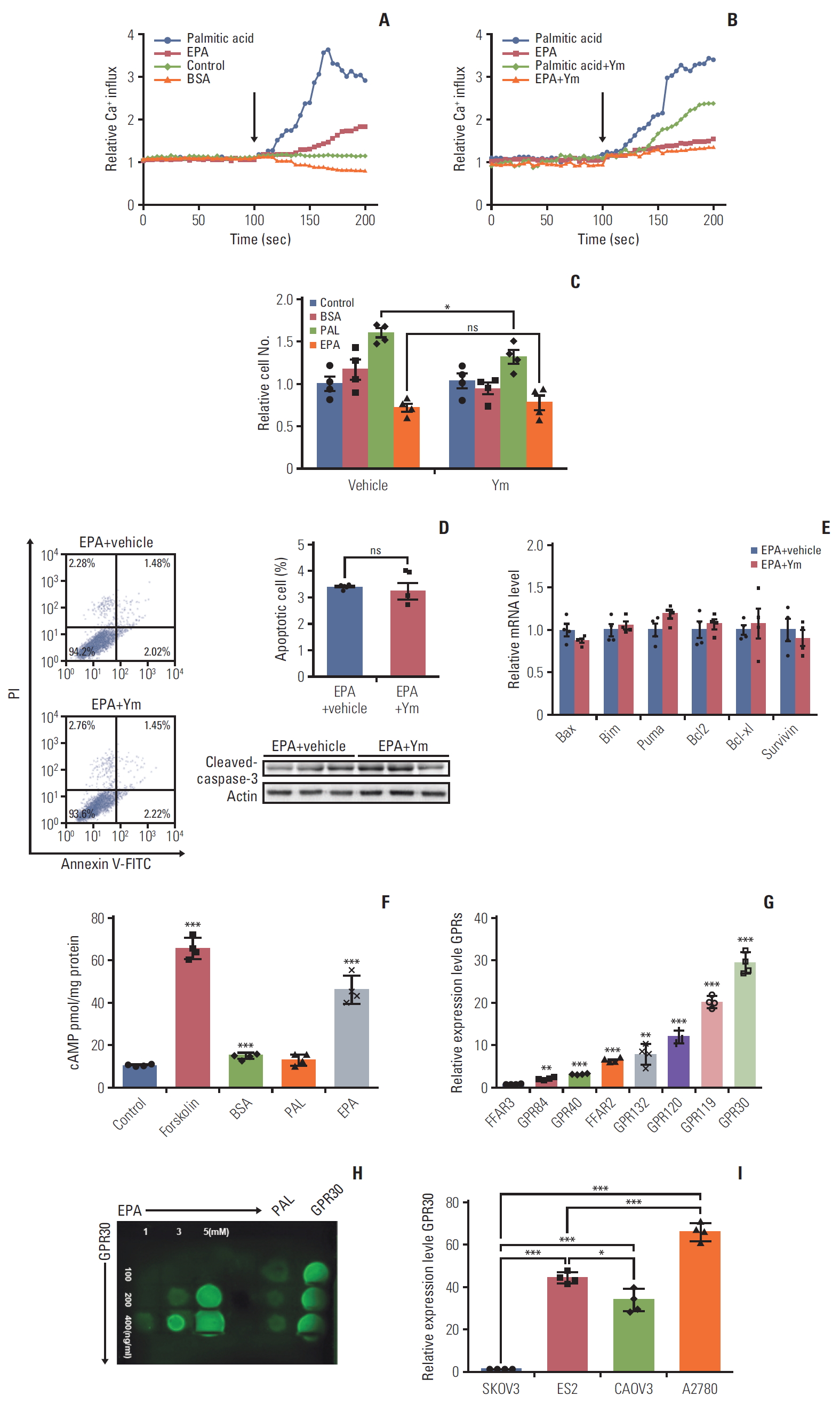
Previous findings suggest the receptors of long-chain FAs are GPR40 and GPR120 [20], and these GPR agonists could induce cell proliferation [21]. There are four subfamilies of α subunits of G proteins: Gαs, Gαi, Gαq, and Gα12. GPR40 and GPR120 couple to the αq subfamily of G proteins, which can be inhibited by YM254890 [10], increasing the downstream effector Ca2+ [22]. To determine whether GPR40 and GPR120 mediate the effects of palmitic acid and EPA in ES2 cells, we measured changes in [Ca2+] in cells following palmitic acid or EPA treatments in the absence and presence of the Gq inhibitor YM254890, which can completely block the Gq pathways [10]. The modest increase in Ca2+ that we detected could be attributed to Gβγ, which could have activated PLCβ on the cell membrane and finally cause Ca2+ release from the endoplasmic reticulum. Without YM254890 treatment, palmitic acid and EPA activated [Ca2+] increases of 4- and 1.5-fold, respectively (Fig. 2A). Interestingly, with YM254890 treatment, the increase in [Ca2+] by EPA did not change, while the increase in [Ca2+] by palmitic acid was inhibited (Fig. 2B). These results indicate that palmitic acid functions mainly through the Gq pathway, while EPA might have its effects through other Gα pathways.
Similarly, we studied the effect of YM254890 on palmitic acid proliferation as well as the EPA antiproliferative effect in ES2 cells. As shown in Fig. 2C, YM254890 decreased palmitic acid-induced proliferation by 15% (Fig. 2C). However, YM254890 did not affect EPA-induced antiproliferation or proapoptosis effects (Fig. 2C and D). Consistent with the data above, the expression of proapoptotic genes, antiapoptotic genes, and cleaved caspase-3 as influenced by EPA was not changed by YM254890 (Fig. 2E). The results above suggest that GPR40 or GPR120, whose subunit is Gαq, is not involved in the anticancer effect of EPA in ES2 cells. Unlike the Gαq family, whose downstream effect is increasing [Ca2+], Gαs increases but Gαi decreases the level of cAMP [22]. To identify which Gα subunit functioned when ES2 cells were treated with EPA, we first measured the production of cAMP in ES2 cells treated with palmitic acid or EPA. The results showed that EPA but not palmitic acid increased cAMP compared to vehicle (Fig. 2F), suggesting that the EPA receptor is coupled to Gαs protein.
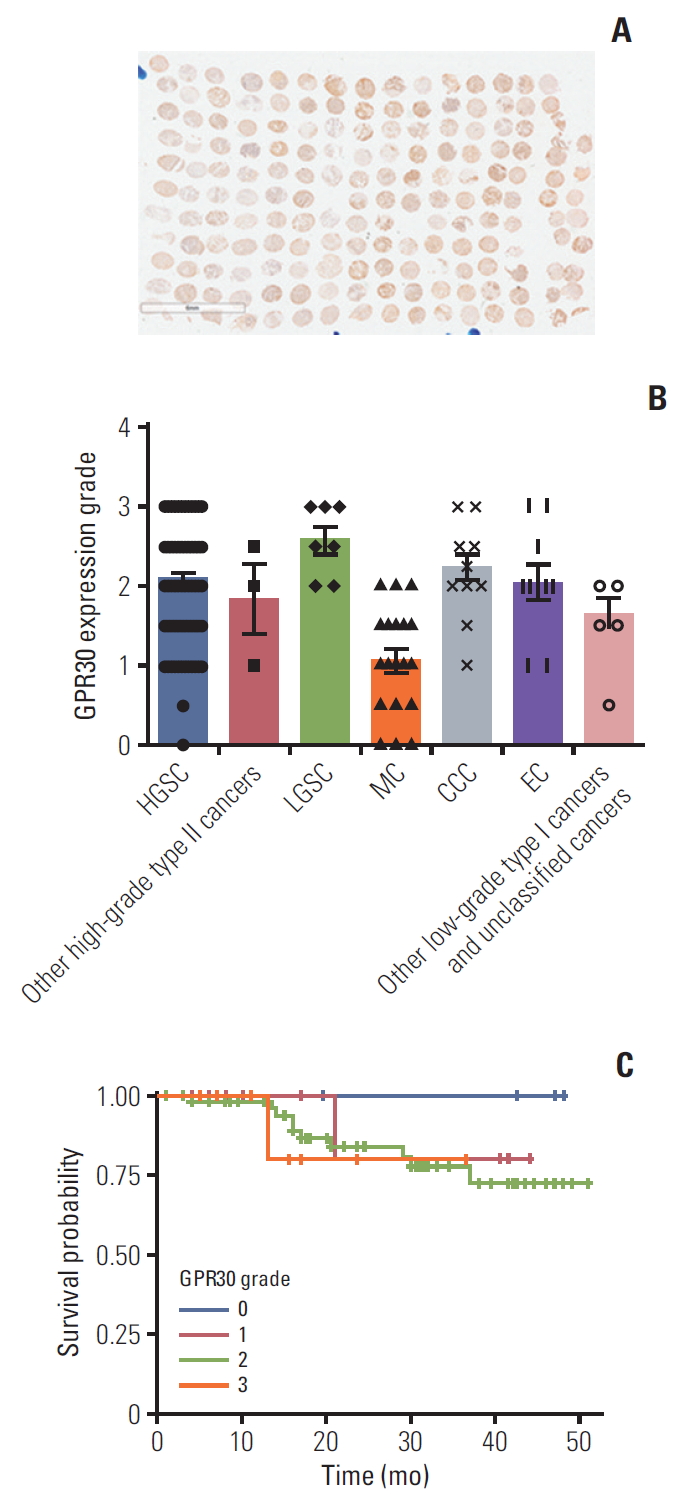
Based on the above results, we hypothesized that there exists some GPR whose Gαs subunit plays a dominant role in the EPA effect in ES2 cells. Thus, we detected various GPR proteins expression levels in ES2 cells. Quantitative PCR analysis showed that GPR30, coupled to Gαs protein [23], was significantly higher than other GPRs (Fig. 2G). We also detected and graded expression of GPR30 in patients with various ovarian cancer, including high-grade serous carcinomas, low-grade serous carcinomas (LGSC), mucinous carcinomas (MC), clear cell carcinomas (CCC), and endometrioid adenocarcinoma. The immunohistochemistry results showed that GPR30 was widely expressed in patients with various ovarian cancers, highly in LGSC while lowly in MC (Fig. 3A and B). Furthermore, the correlation analysis of GPR30 expression level with overall survival of ovarian cancers patients showed that patients with higher GPR30 expression level (grade 3) showed shorter survival time, while patients with lower GPR30 expression level (grade 0) showed longer survival time (Fig. 3C, S1-S3 Table).To further verify whether EPA was a ligand of GPR30 in ES2 cells, we used a modified protein lipid overlay assay [14]. As shown in Fig. 2H, the interaction between EPA and GPR30 existed and was dose-dependent. Moreover, the relative expression level of GPR30 was markedly lower in SKOV3 cells compared with ES2 cells, A2780 cells, and CAOV3 cells, indicating that GPR30 functions differently in different cell lines (Fig. 2I).
3. GPR30 mediates the EPA effect in ES2 cells
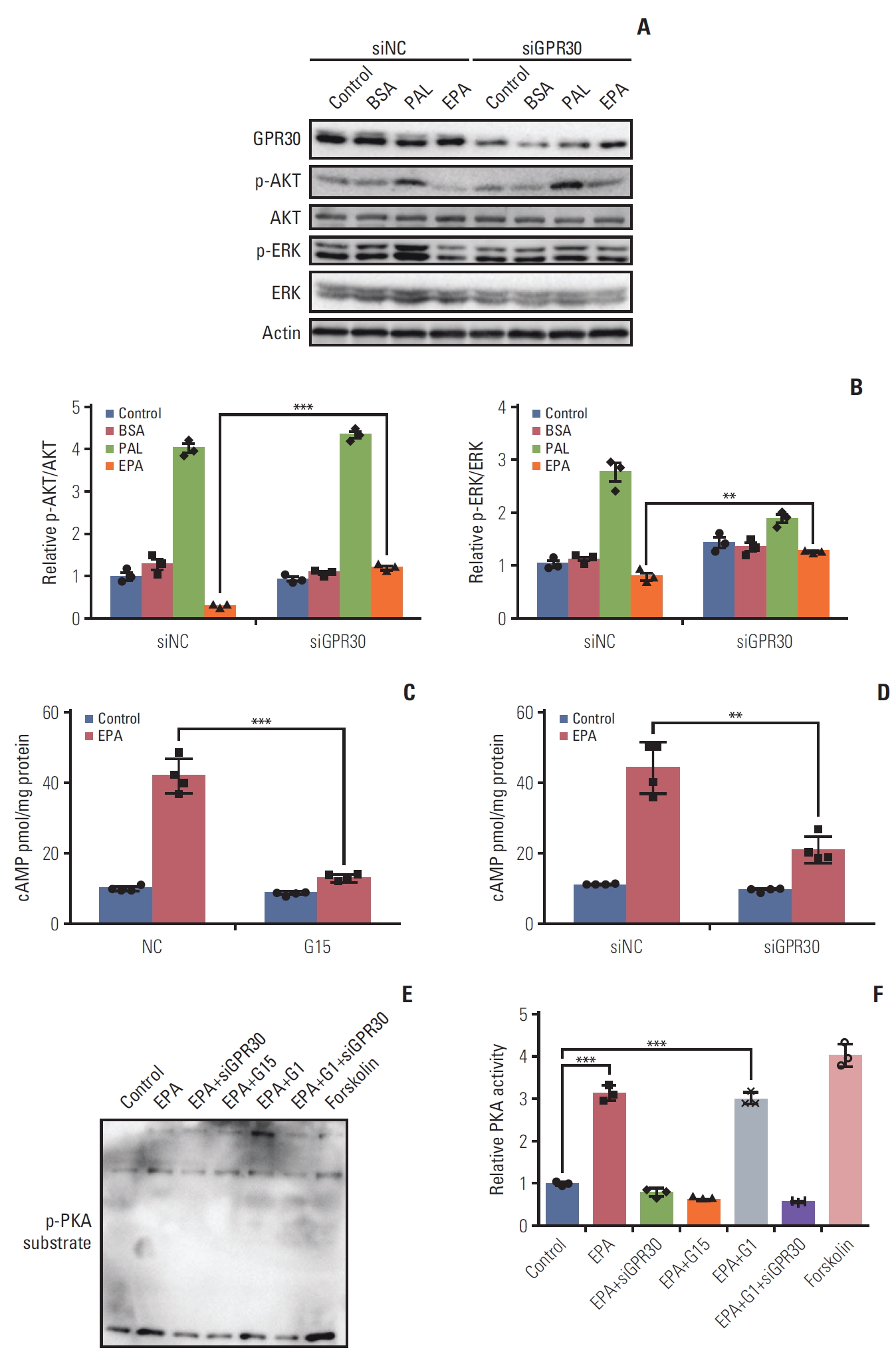
To determine whether GPR30 mediates the effect of EPA in ES2 cells, we knocked down GPR30 using RNAi and found that GPR30 expression was reduced by 80% without changes in GPR40 or GPR120 (Fig. 4A). The decrease in cell number following EPA treatment was significantly reversed in ES2 cells treated with GPR30-specific siRNA (Fig. 4B). Similarly, we used G15, the specific antagonist of GPR30, to test the EPA effect in ES2 cells. As shown in Fig. 4C, the inhibitory effect of EPA was again impaired. The siRNA and inhibitor results indicated that EPA-induced antiproliferation in ES2 cells is mediated through GPR30.
Furthermore, we knocked down GPR30 to examine its effect on the EPA proapoptotic action in ES2 cells. The number of apoptotic cells dramatically declined when ES2 cells were treated with GPR30 siRNA compared with negative-control siRNA (Fig. 4D). Additionally, the expression of apoptotic genes as influenced by EPA was reversed when GPR30 was knocked down (Fig. 4D and E). However, the proapoptotic effect of EPA on ES2 cells could not be reversed when GPR119 was knocked down (S5 Fig.), indicating GPR119, another Gαs coupled receptor, did not play an essential role in mediating the anticancer effect of EPA in ES2 cells. Altogether, the above results establish that GPR30 is essential in mediating the effect of EPA in ES2 cells.
4. EPA inhibits cell proliferation through the GPR30-cAMP-PKA signaling pathway in ES2 cells
The phosphoinositide 3-kinase (PI3K)/AKT signaling pathway is the most common pathway related to cancer proliferation [24]. In recent years, numerous reports have demonstrated that EPA is implicated in several signaling pathways, such as RAS/ERK/C/EBPβ and nuclear factor κB, contributing to tumor development and inflammation [25]. To evaluate whether EPA affects cell proliferation through these cellular response pathways, we investigated phosphorylated AKT and ERK1/2 in ES2 cells treated with or without EPA. Interestingly, palmitic acid stimulated AKT and ERK1/2 phosphorylation, while EPA inhibited their phosphorylation after 24 hours of treatment (Fig. 5A). To further determine whe-ther GPR30 was involved in the activation of AKT and ERK1/2, we knocked down GPR30 as mentioned above. The phosphorylation levels of AKT and ERK1/2 in response to palmitic acid did not obviously change, and that in response to EPA was dramatically increased when ES2 cells were treated with GPR30 siRNA (Fig. 5A and B). These findings suggest that the development of signaling pathways via GPR30 includes PI3K-AKT and ERK.
The results in Figs. 2 and 4 show that EPA inhibits ES2 cell growth through GPR30. Due to the inhibitory effect on cancer cell growth by Gαs-cAMP-PKA signaling activation [26], the Gαs-cAMP-PKA signaling pathway may be affected by EPA via GPR30 in ES2 cells. Thus, we knocked down GPR30 in ES2 cells by GPR30 siRNA and G15 as described above. The cAMP production promoted by EPA was attenuated following downregulation of GPR30 (Fig. 5C and D). Additionally, EPA increased PKA activity compared to the treatment vehicle, as detected by the phospho-(Ser/Thr) PKA substrates. Notably, G1, the specific agonist of GPR30, also increased PKA activity. Moreover, PKA activity stimulated by EPA was suppressed by GPR30 knockdown and G15. However, PKA activity stimulated by EPA, G1, and siGPR30 did not change compared to its activity stimulated by EPA and siGPR30 (Fig. 5E and F). These results indicate that GPR30-cAMP-PKA signaling is involved in the EPA effect in ES2 cells.
5. EPA blocks tumor growth via GPR30 in mouse xenografts
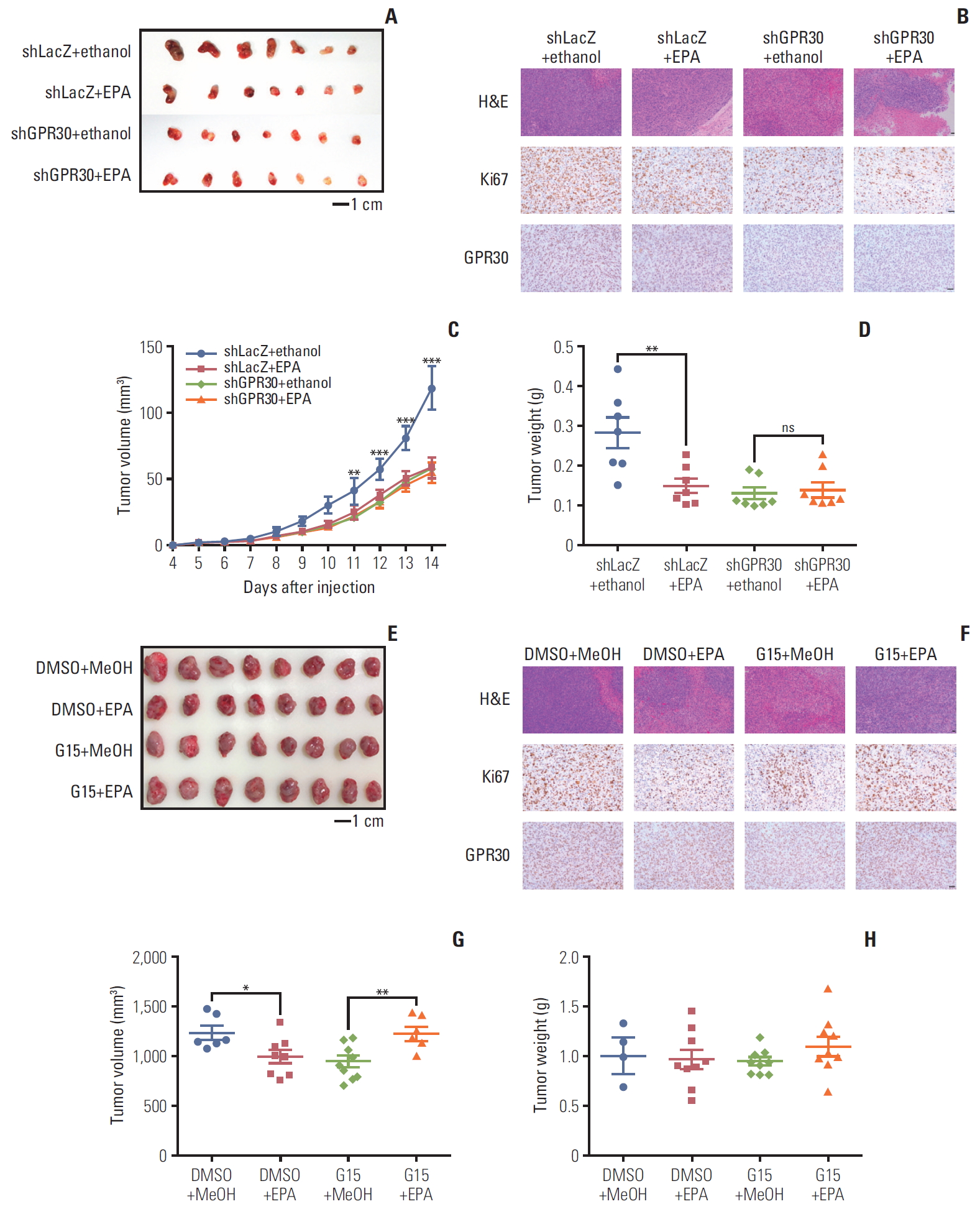
To confirm the above observations, we also conducted a mouse xenograft model to further verify the role of GPR30 in ES2 cells. In accordance with previous findings, EPA alone inhibited tumor growth significantly (Fig. 6A and E). To further confirm the function of GPR30, we knocked down GPR30 using adenovirus in a mouse xenograft model. As shown in Fig. 6A, EPA dramatically decreased tumor growth, while EPA was unable to inhibit tumor growth when GPR30 was knocked down. hematoxylin and eosin (H&E) staining and labeling for the cancer proliferation marker Ki67 also showed that EPA lost its inhibitory function when ES2 cells were treated with GPR30 shRNA (Fig. 6B). The change in tumor volume and weight supported the above results (Fig. 6C and D). Interestingly, we found that the deletion of GPR30 in ES2 cells decreased tumor growth in mice, suggesting that GPR30 is associated with tumor development. Therefore, we antagonized GPR30 activity by treatment with GPR30 antagonist G15 in the mouse xenograft model. Although the tumor weight had no significant change due to individual variation (Fig. 6H), the tumor size and H&E staining showed that EPA or G15 alone both inhibited cell proliferation significantly (Fig. 6E-G). The combination of EPA and G15 was unable to inhibit tumor growth, as G15 inhibited antitumor effect of EPA by impairing GPR30 activity. These in vivo results also suggest that EPA inhibits tumor growth via GPR30 in human ovarian clear cancer cells.
Discussion
Extensive research implies that dysregulation of lipid metabolism is correlated with ovarian cancer progression [27]. EPA, an n-3 polyunsaturated FA, has anticancer effects in many cancer cells, such as colorectal cancer [28], breast cancer [3], pancreatic cancer [28], and ovarian cancer [5]. In our study, EPA-induced apoptosis in ES2 OCCC cells following induction of antiproliferation through GPR30, a novel EPA receptor. Additionally, EPA stimulated the activation of caspase-3, blunted the activation of AKT and ERK1/2 and functioned through the GPR30-cAMP-PKA signaling pathway.
Classical free fatty acid receptors, such as GPR40, and GPR120, might also mediate the function of EPA in ovarian cancer cells. Since Gαq is the α subunit of both GPR40 and GPR120, whose activation leads to a rapid increase in Ca2+, we detected the Ca2+ concentration after adding EPA, and an approximately 1.5-fold increase was observed. Importantly, YM254890, a specific inhibitor of the Gαq unit, did not inhibit the increase in Ca2+ caused by EPA, suggesting that neither GPR40 nor GPR120 is the specific receptor of EPA. We found a novel EPA receptor, GPR30, in ovarian cancer cells, confirmed by a modified protein lipid assay [14], thus broadening the concept of cancer metabolism. GPR30, which was once thought to be an orphan receptor, has been implicated in both rapid and transcriptional events in response to estrogen. Ligands of GPR30 are mainly steroids and some synthetic estrogen-receptor ligands, and the pro-proliferation effects of E2 in hormone-related tumors are well known. When we blocked GPR30 expression by shRNA in vivo, we also blocked the pro-proliferation effects of E2 because of the lack of ERα and ERβ in ES2 cells. Therefore, the volume and weight of these tumors were significantly decreased, as shown in Fig. 6D. Above all, we first proved that besides steroids, EPA is also a ligand for GPR30.
Oxidative stress has been reported to affect cancer cell development. For example, reactive oxygen species (ROS) participate in cancer cell progression and proliferation, cell apoptosis, and energy metabolism [29]. Previous reports showed that EPA mainly causes ROS-induced apoptosis [28]. The cell death, which mainly occurs in the late apoptosis phase, is due to the intracellular ROS-induced caspase-8 activation [30]. Other evidence has demonstrated that EPA also induces autophagic cell death [28]. Here, we show that EPA increased the activation of caspase-3 and stimulated the expression of apoptotic genes. We found that EPA promoted a less antiproliferative signal in ES2 cells but an insensitive response in SKOV3 cells treated with GPR30 siRNA. Considering that different types of ovarian cancer have distinctive morphologic and molecular-genetic features, GPR30 may be coupled to G proteins more tightly in ES2 cells than in SKOV3 cells. Another reason may be that the enrichment of GPR30 protein varies between ovarian cancer cells and that SKOV3 cells have a low expression level of GPR30 (Fig. 2I). Besides, EPA exists pro-proliferation effect on CAOV3 and A2780 cell lines, indicating the antitumor effect of EPA may be cell-line specific event. Therefore, further investigation about the effect of EPA on other CCC cells should be required.
Our observations suggest that in ES2 OCCC cells, EPA functions through the GPR30-Gαs-cAMP-PKA signaling pathway. Originally, phosphorylation of ERK1/2 is increased by Gβγ, which is dissociated from Gα. Previous work also demonstrated that PKA stimulated by the α subunit of GPR30 could decrease the phosphorylation of ERK1/2 [26]. We think that crosstalk between them may account for the decreased phosphorylation level in our study. Further investigation will elucidate the specific functions of signaling pathways activated by EPA, including autophagy, metabolism, cell cycle.
Overall, our study provides evidence that GPR30 is a novel receptor of EPA in ES2 cells that mediates its anticancer effects. EPA might inhibit proliferation by inducing apoptosis. Additionally, EPA might influence the GPR30-ERK or GPR30-AKT signaling pathway. In our future work, we will examine the mechanism of the effects that EPA has on other backgrounds. In addition, because other researchers have provided evidence that EPA can improve the sensitivity to chemotherapy [2], we wondered whether EPA could improve the chemotherapy resistance of OCCC through GPR30. Answering these questions could provide a theoretical basis for the clinical treatment of OCCC.