Introduction
Around 7% of malignant neoplasms occur in the kidney; these include Wilms tumor (WT; also known as nephroblastoma), clear cell sarcoma of the kidney, malignant rhabdoid tumor, and renal cell carcinoma [1,2]. WT is the most common primary kidney tumor in children and is treated with a multimodal approach that consists of surgery, radiation therapy, and anticancer drugs [3]. Based on tumor stage and pathological findings of upfront nephrectomy, the National Wilms Tumor Study Group (NWTSG) recommends treating WT according to the risk of relapse [4]. In contrast, the International Society of Pediatric Oncology advocates upfront chemotherapy before nephrectomy, with the intensity of subsequent therapies adjusted according to tumor response [5]. Attempts to treat WT patients according to the risk of tumor recurrence using prognostic markers has led to improvement in outcome over the past 40 years, such that the long-term survival rate is now 90% [6].
Tumor suppressor genes contribute to tumorigenesis by disabling the function of both normal alleles of a gene [7,8]. Loss of heterozygosity (LOH) at a chromosomal locus comprising a tumor suppressor gene results in functional defects that can lead to pediatric WT [7,9]. It was recently reported that LOH at chromosome 1p and/or 16q is associated with high recurrence and mortality rates in WT patients. The National Wilms Tumor Study (NWTS)-5 reported that tumor-specific LOH at 1p and 16q was associated with a higher risk of relapse and death in favorable histology Wilms tumor (FHWT), and treatment intensity was increased in these patients [6].
However, different investigators have reported variable findings regarding the prognostic value of LOH at 1p and 16q [10-12]: some have shown that LOH at both loci predicts survival, while others have found that only one or the other affects prognosis [13]. These discrepancies could be due to several causes, not only differences in the study populations, but also differences in ethnic variation. The present study investigated the prognostic significance of LOH at 1p and 16q in a Korean pediatric FHWT cohort.
Materials and Methods
1. Patients and treatment
The study protocol was approved by the WT committee of the Korean Society of Pediatric Hematology Oncology Group (K-PHOG), which analyzed patient information and specimens from the four participating hospitals, each of which passed Institutional Review Board deliberation, filled out the case report form (CRF), and sent five-panel slides for each patient to the committee for retrospective review. All patints included in this study were diagnosed as FHWT between 1996 and 2016; examined by chest X-ray, abdominal sonography, computed tomography, or magnetic resonance imaging; staged according to the NWTSG staging system; and treated according to National Wilms’ Tumor Study 4 (NWTS-4) guidelines [6]. One institution modified the NWTS-4 treatment guideline; initial fine needle biopsy and delayed nephrectomy was done at 6 to 9 weeks if the largest diameter of the tumor is greater than 8 cm (modified NWTS). The CRF included data on the patient’s age, sex, histological findings, staging at the time of diagnosis, surgery, radiation, chemotherapy, relapse, and survival.
Upfront nephrectomy was initially recommended for all patients. However, chemotherapy was given prior to nephrectomy if the tumor was deemed unresectable by the surgeon or if the surgery was life-threatening, the tumor thrombus extended into the inferior vena cava above the level of the hepatic veins, or the disease was bilateral by Doppler sonography. Stage I-II patients received two drugs including vincristine and actinomycin D for 18 weeks without radiotherapy, and stage III-IV patients were treated with a threedrug regimen consisting of vincristine, actinomycin, and doxorubicin for 24 weeks in addition to receiving radiotherapy. Stage V patients were treated with nephron-sparing surgery and chemotherapy or delayed nephrectomy after preoperative chemotherapy and then postoperative chemotherapy.
2. Histopathological examination and DNA extraction for microsatellite analysis
Paraffin-embedded kidney specimens containing normal and tumor tissues that were cut into 5-μm sections and mounted on glass slides (five per patient) were sent from participating institutions to that of the principal investigator. Pathological diagnoses were reviewed at a central location and tumor cells were microdissected under a microscope from hematoxylin and eosin-stained sections. DNA was extracted from tumor and non-tumor sections for microsatellite analysis according to standard procedures. Two tissue sections from each case were transferred using a disposable razor blade to 900 μL of xylene in a 1.5-mL microcentrifuge tube. Equal volumes of 100% and 70% ethanol were added and the samples were pelleted (10 minutes in a microcentrifuge), dried at 56°C, and incubated overnight in cell lysis buffer (cat. No. 1045723, Qiagen, Valencia, CA) with 1.5 μL of proteinase K (41 μg/mg). The proteinase K was inactivated by heating the samples at 56°C for 30-60 minutes; 100 μL of protein precipitation solution (cat. No. 1045701, Qiagen) was added to each sample, followed by centrifugation for 5 minutes. The supernatant was removed and isopropanol was added to precipitate the DNA, which was washed with 70% ethanol and transferred to a new 1.5-mL tube.
3. Detection of LOH at 1p and 16q
To detect LOH at 1p and 16q, 16 polymorphic microsatellite markers were selected based on a previous study [6]. Forward primers were labeled with four different fluorescent dyes (VIC, 6FAM, PET, or NED) (Table 1). Polymerase chain reaction (PCR) amplification was performed on tumor and normal tissue samples in triplicate using the Qiagen Multiplex PCR Plus Kit (Hilden, Germany) under the following conditions: 95°C for 10 minutes; five cycles of 95°C for 20 seconds, 60°C for 90 seconds (−1°C in each cycle), and 72°C for 1 minute; 30 cycles of 95°C for 20 seconds, 55°C for 90 seconds, and 72°C for 1 minute; and 60°C for 30 minutes. PCR products were examined using a 3500xL Dx Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA) and LOH was detected using GeneMapper v.4.1 software (Thermo Fisher Scientific) according to the manufacturer’s instructions.
4. Statistical analysis
Event-free survival (EFS) refers to the time from diagnosis to the first occurrence of progression, relapse after response or death from any cause. Overall survival (OS) was defined as the time from diagnosis to the day of death or last follow up. EFS and OS were calculated with the Kaplan-Meier method, and the log-rank test and the Cox proportional hazards model were used for uni- and multivariate analyses, respectively. The chi-square test was used to compare the contingency tables. Two-sided p-values < 0.05 were considered statistically significant.
5. Ethical statement
This study was performed in accordance with the ethical guidelines of the “World Medical Association Declaration of Helsinki-Ethical Principles for Medical Research Involving Human Subjects.” This study was approved by the Institutional Review Board of participating institutions and Ajou University Hospital (IRB No. AJIRB-BMR-OBS-16-143). Written informed consent was waived from the IRB.
Results
A total of 101 FHWT patients were enrolled in the study. The median age was 2.8 years (range, 0.3 to 10.4 years). The male-to-female ratio (48:53) was 1.04. The characteristics of the study population are summarized in Table 2. Among the patients, 43 (42.6%) and 52 (51.5%) had tumors in the right and left kidneys, respectively, whereas six (5.9%) had tumors in both kidneys. Eleven patients showed distant metastasis to the lung. Upfront nephrectomy was performed in 54 patients and delayed nephrectomy in 47; the latter was performed after preoperative chemotherapy since 37 cases were deemed inoperable by the surgeon, with six cases of bilateral disease and four in which the tumor thrombus extended into the inferior vena cava above the level of the hepatic veins. Forty patients received radiotherapy.
The status of LOH at 1p and 16q in the patients is shown in Table 3. There were 19 patients (18.8%) with LOH at 1p and 16q respectively, including five exhibiting LOH at both loci. The frequency of LOH at 1p was higher among younger patients (p=0.049), especially those under 4 years of age. There was no difference in LOH prevalence according to tumor stage (Table 3).
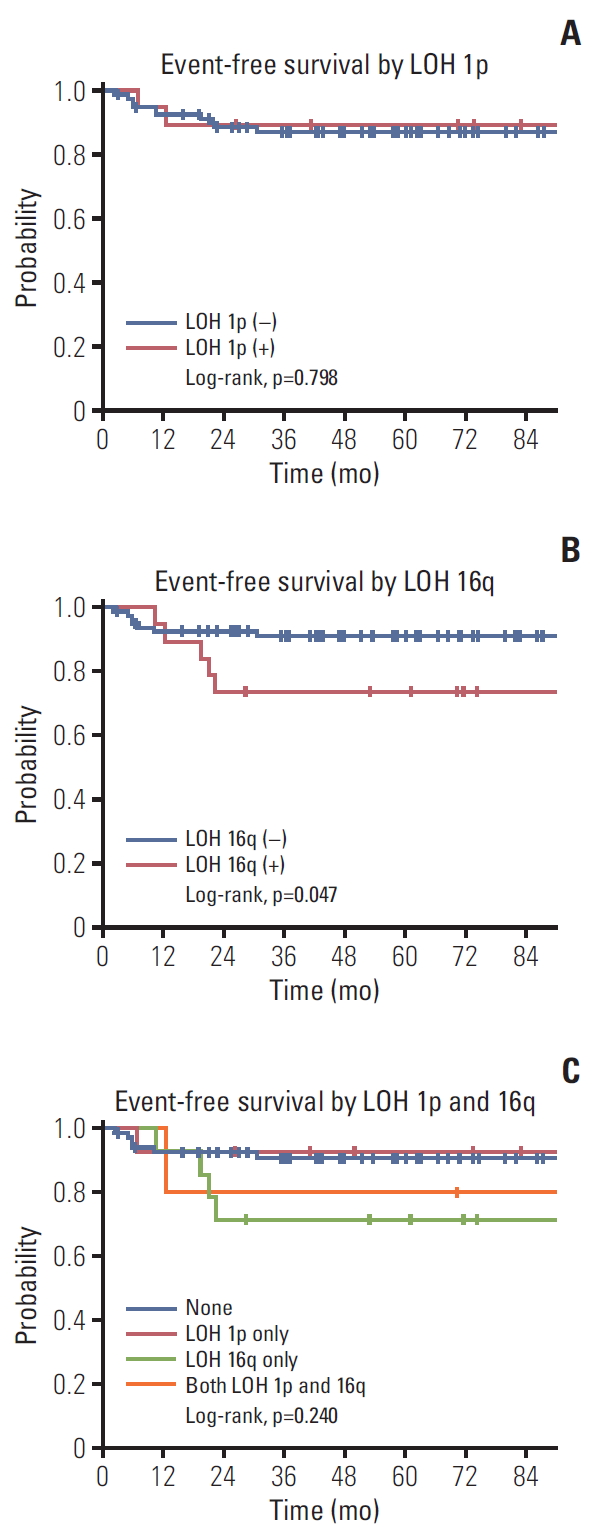
Among the 101 patients, there were 12 recurrences and two deaths. The 3-year OS and EFS of all patients were 98.0% and 87.6%, respectively. The 3-year EFS rate was 89.5% in patients with and 87.2% in those without LOH at 1p (p=0.798 [log-rank test]) (Fig. 1A). The 3-year EFS rate was lower in patients with LOH than in those without LOH at 16q (73.7% vs. 91.1%, p=0.047 [log-rank test]) (Fig. 1B). Patients with LOH at both 1p and 16q did not show a lower EFS rate than those in the other groups (Fig. 1C).
A multivariate analysis showed that LOH at 16q was a significant negative prognostic factor for EFS (hazard ratio, 3.95; p=0.037), which was not true of LOH at 1p (hazard ratio, 0.83; p=0.817) (Table 4).
Discussion
There has been significant progress in the treatment of FHWT over the past 30 years, and the survival rate is now about 90% [14]. This excellent outcome is the result of a multimodal approach that combines surgery, chemotherapy, and radiation therapy according to tumor stage and histology. Additionally, tumors at high risk of recurrence are treated more aggressively than those at low risk, such that overtreatment can be avoided in the latter cases [15]. Recently, efforts have been made to identify new molecular markers that can be used to predict tumor recurrence by stratifying high- and low-risk patients, which can ultimately prolong survival by reducing long-term treatment sequelae [7,9]. There have been many reports of LOH in relation to the risk of recurrence of WT, for instance at 1p, 11q, 16q, and 22q [10,13,16]. Of these, LOH at 1p and LOH 16q are the most widely investigated and were the focus of the present work.
In our study, 19 patients each out of 101 (18.9%) had LOH at 1p and 16q. We compared these rates to the ones reported in other studies (Table 5). Of the 232 WT patients enrolled in NWTS-3 and -4, 21 patients (12%) had LOH at 1p and 35 (17%) at 16q [10]. In NWTS-5, 196 out of 1,727 FHWT patients (11.3%) had LOH at 1p and 301 (17.4%) had LOH at 16q [4]. Of the 426 FHWT patients enrolled in the UKW 1-3 trials, the prevalence of LOH at 1p and 16q was 10.3% and 17.4%, respectively [11]; among the 125 patients who participated in the AIEOP-TW-2003 trial, the rates were 19% and 15%, respectively [13]; and in an Egyptian FHWT cohort (n=100), the rates were 16% and 25%, respectively [12]. Thus, with the exception of one report [12], the prevalence of LOH at 1p and 16q is about 10%-19% and 14%-20%, respectively, whereas the proportion of patients with both alterations is between 2.6% and 5% (i.e., 4.6% [4], 2.6% [11], and 4% [13]). Five out of 101 patients in our study had LOH at both 1p and 16q, which is comparable to the rates in these other studies.
The frequencies of LOH at 1p and 16q are higher among FHWT patients over 4 years old [4], and lower in patients younger than 2 years old [12]. One study found that the frequency LOH at 1p but not at 16q increased after 4 years of age [11]. Others have shown a low prevalence of LOH at 1p but not at 16q in patients under 2 years old [13]. In our study, the rate of LOH at 1p was higher among younger FHWT patients (p=0.045), especially in those under 4 years old (Table 3). These same age-group differences in the frequency of LOHs appearing among the reports may represent interracial biological differences in WTs. It was reported that the frequency of LOH at 1p or 16q increases with tumor stage [12]; however, in most other studies [6,11,13] including ours, there was no association between the frequency of these alterations and tumor stage.
Several studies have investigated whether LOH at 1p and/ or 16q can predict FHWT recurrence and patient survival (Table 5). NWTS-5 is the largest of these studies to date; it was prospective in nature and used stage-specific treatment, and showed that LOH at both 1p and 16q loci was associated with worse prognosis [4]. In NWTS-3 and -4, which preceded NWTS-5, LOH at 16q was associated with 3.3- and 12-fold higher rates of relapse and mortality, but LOH at 1p tended to increase recurrence and mortality rates without statistically significant [2,10]. In this study, it is thought that the preceding NWTS-3,4 study was a statistical difference caused by the smaller sample sizes than the NWTS-5 study.
However, it is thought that there would be another reason for different results in determining LOH(s) at 1q and/or 16q as prognostic factor(s). In a study of 125 children with FHWT treated with the AIEOP-TW-2003 protocol which was conducted in Italy from 2003 to 2008, LOH at 1p was associated with worse disease-free survival [13]. Meanwhile, in the UKW 1-3 trials conducted in the United Kingdom with a relatively large number of subjects, 426 FHWT patients, LOH at 16q but not at 1p was also associated with a higher risk of relapse and death [11]. In a study of Egyptian FHWT patients, LOH at 16q alone or in combination with LOH at 1p had a higher recurrence rate and lower 3-year EFS [12]. It can be inferred that genetic variability of the WT may have racial differences to produce different outcomes for each country that conducted the study. In our study, EFS was lower in patients with LOH at 1p only or at both 1p and 16q compared with those with neither alteration; this was not statistically significant, possibly due to our small sample size. Nonetheless, our results show that only LOH 16q negatively affects prognosis, which is consistent with previous findings [4,10-12]. This result is thought to reflect racial differences, as stated earlier in the study conducted in United Kingdom, Egypt, and Italy.
Only cases of LOH at both 1p and 16q in FHWT stages I-IV are stratified into poor prognosis groups and receive additional intensive chemotherapy, although both types of alteration show a low prevalence of 4% [6]. Thus, the absolute number of patients who would benefit from clinical decisions made based on the identification of LOH may be relatively small if they were classified as high risk and given intensive chemotherapy [13]. In our study, five patients among the 19 with LOH at 16q experienced recurrence; assuming that this group be received more aggressive treatment in the future study, the remaining 14 will be likely overtreated. Therefore, it is important that patients are stratified with higher statistical power and treated in combination with other risk factors rather than classified as a recurrent risk group based solely on detection of LOH at 16q [9].
The limitation of our study is that although it was a nationwide study conducted by K-PHOG the sample size was relatively small, this is especially true for LOH at 1p that is statistically insignificant. Second, the study was retrospective and therefore did not have a uniform staging system and treatment strategy. Therefore, the next study needs a prospective study with a uniformed staging system and treatment strategy.
This study is meaningful in that it is the first to identify LOH at 16q as a significant negative prognostic factor affecting outcome in Korean pediatric FHWT patients. However, this study is limited to its small sample size, large-scale multicenter trials are warranted to investigate the prognostic value of LOH at 1p and 16q in Korean children with FHWT.