Introduction
Pancreatic cancer (PC) is one of the most lethal diseases worldwide, and there is an urgent need to develop effective therapies for this disease [1,2]. Recently, genomic analyses have revealed that many cancer susceptibility genes are frequently mutated in PC, including KRAS (92% of cases) and TP53 (78%). In addition, germline and somatic mutations in genes encoding breast cancer 1 and 2 (BRCA1, BRCA2), ataxia telangiectasia mutated (ATM) and partner and localizer of BRCA2 (PALB2) are present in PC patients with frequencies of 5% (germline mutations) and 12% (somatic mutations) [1]. These genes play critical roles in the DNA damage response (DDR), which signals the presence of strand breaks and other forms of DNA damage and coordinates their repair. Paradoxically, mutations in genes that compromise the DDR can both cause and protect against cancer. On the one hand, defects in the DDR can lead to genomic instability and the accumulation of mutations that increase the probability of cancer. On the other hand, DDR pathway dysfunction can render tumor cells susceptible to chemotherapeutic agents that damage DNA and/or impair alternative DDR pathways [3]. Thus, targeting of specific molecules in the DDR is a potential strategy for the development of new drugs for cancers with urgent unmet needs, including PC.
The cellular response to DNA damage, including single-stranded or double-stranded DNA breaks, is controlled by a network of proteins that include damage-sensing proteins such as poly(ADP-ribose) polymerase (PARP); transducers such as the kinases ATM and ataxia telangiectasia and Rad3 related (ATR); and effectors such as the nuclear kinase WEE1, which is a key protein in cell cycle checkpoint control and inhibits entry into mitosis [3]. Ultimately, the DDR results in one of three outcomes for the cell: successful DNA repair, cell cycle arrest, or apoptosis.
Therapeutic targeting of the DDR pathway has been examined in diverse tumor types [3]. Notably, inhibition of PARP, ATM, ATR, or WEE1 has been shown to abrogate DNA repair via homologous recombination (HR) in many cancers with a genetically defective DDR, thus leading to synthetic lethality [4-7]. WEE1 acts as a gatekeeper of the G2/M cell cycle checkpoint, and its activity increases during the S and G2 phases; thus, WEE1 inhibition can induce growth arrest in S phase [8]. In contrast, cancer cells expressing mutant TP53 lack a functional G1 checkpoint, and DNA damage must be repaired during the G2/M transition. Given that TP53 is often mutated in PC [8], PC is the good candidate for the development of the DDR-acting agents.
The tumor microenvironment plays a critical role in cancer progression [9]. PC is unique compared with other tumor types in being surrounded by strong stroma. Abundant immunosuppressive cells reside in the tumor microenvironment, including regulatory T cells, myeloid-derived suppressor cells (MDSCs), M2-type macrophages, and cancer-associated fibroblasts (CAFs) [9,10]. Recruitment of these cells establishes a barrier to the anti-tumor immune response [9,10]. In addition, signaling via the chemokine receptor CXCR2 can drive PC growth by recruiting MDSCs and tumor-associated neutrophils and by enhancing the metastatic process [10]. Programmed cell death ligand 1 (PD-L1) is a negative regulator of the immune response and acts by binding to its receptor programmed cell death 1 (PD-1) on T cells, which inactivates the cells and thus allows the tumor to escape immune surveillance [11]. Data from genomic analyses indicate that immunogenic subtype of PC, which exhibits high levels of PD-L1, cytotoxic T-lymphocyte-associated protein 4 and CXCR2 among several subtypes [1]. Notably, the chemokine-like factor-like MARVEL transmembrane domain containing family member 6 (CMTM6) has been suggested as one of the mechanisms of regulation of PD-L1 through preventing PD-L1 degradation by lysosome [12].
Increasing evidence suggests the existence of crosstalk between the DDR signaling network and immune pathways [13,14]. For example, recent studies have demonstrated that the DDR regulates PD-L1 expression in cancer cells via a pathway involving activation of signal transducer and activator of transcription (STAT) signaling and inactivation of glycogen synthase kinase-3β (GSK-3β) [15,16]. However, such interactions between the DDR and immune signaling have not yet been studied in PC.
Here, we evaluated the anti-tumor effects of targeting the DDR using a WEE1 kinase inhibitor (AZD1775) and an ATM kinase inhibitor (AZD0156) in PC cells in vitro and in a mouse xenograft model. We also examined the expression and activation of a number of DDR-related and immune signalingrelated molecules to identify potential crosstalk between the DDR and immune system in PC.
Materials and Methods
1. Human cell lines and reagents
Ten human PC cell lines were employed in this study: AsPC-1, Capan-1, Capan-2, MIA PaCa-2, PANC-1, SNU213, SNU324, and SNU410 were purchased from the Korean Cell Line Bank (Seoul, Korea), and SNU2913 and SNU2918, patient-derived cell lines, were successfully established from patient. Cells were cultured in medium (MIA PaCa-2 and PANC-1 cells in Dulbecco's modified Eagle's medium, all other cell lines in RPMI-1640, both from Welgen Inc., Gyeongsan, Korea) supplemented with 10% fetal bovine serum and 10 μg/mL gentamicin and were maintained at 37°C in a 5% CO2 atmosphere. The WEE1 inhibitor AZD1775 and ATM inhibitor AZD0156 were kindly provided by AstraZeneca (Macclesfield, UK).
2. PD-L1 expression analysis by flow cytometry
Cells (2×105) were seeded in 60-mm dishes and incubated with AZD1775 and/or AZD0156 for 72 hours. The adherent cells were harvested, resuspended in cell staining buffer (#420201, BioLegend, San Diego, CA), and incubated with anti–PD-L1 antibody (#329708, BioLegend) for 30 minutes at room temperature. Cells were then washed once with the same buffer and analyzed on a FACSCalibur. The results are presented as the means of three independent experiments.
3. Human cytokine array
Cells (1×106) were seeded in 60-mm dishes and exposed to AZD1775 and/or AZD0156 for 24 hours. The cell supernatant was then collected and 500 μL/sample was analyzed using the Proteome Profiler Human Cytokine Array Kit (#ARY-005B, R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Spot intensities were measured using ImageJ software (National Institutes of Health, Betheda, MD).
4. Human phospho-kinase array
Cells (1×106) were seeded in 100-mm dishes and exposed to AZD1775 and/or AZD0156 for 72 hours. The cells were harvested, and lysate samples containing 300 μg of proteins were analyzed using the Proteome Profiler Human PhosphoKinase Array Kit (#ARY003B, R&D Systems) according to the manufacturer’s instructions. Spot intensities measured using ImageJ software.
5. Tumor xenograft experiments
Four-week-old female athymic nude mice were purchased from Orient Bio Inc. (Seongnam, Korea). Capan-1 cells were resuspended at 3×107 cells in 100 μL of phosphate-buffered saline and injected subcutaneously. The tumor volume was calculated using the formula: volume=[(width)2×height]/2. When the tumor volume reached 200 mm3, the mice were randomly assigned to four groups of five mice to receive (1) vehicle (2-hydroxypropyl-β-cyclodextrin solution), (2) AZD1775 once daily at 30 mg/kg for 4 weeks (5 days on/2 days off), (3) AZD0156, as described for (2), or (4) AZD1775 plus AZD0156, as descri-bed for (2). All treatments were administered by oral gavage. Body weights and tumor sizes were measured every other day.
6. Mouse cytokine array
Immediately before sacrifice, the mice were bled and serum samples were prepared. Aliquots of 500 μL were analyzed using the Proteome Profiler Mouse Cytokine Array Kit, Panel A (#ARY006, R&D Systems) according to the manufacturer’s instructions. Spot intensities were measured using ImageJ software.
7. Statistical analysis
Analyses were conducted using SigmaPlot version 10.0 (Systat Software Inc., San Jose, CA). Data are presented as the mean±standard errors. All statistical tests were two-sided. Differences were considered significant if the p-values were < 0.05. Half-maximal inhibitory concentrations (IC50) were calculated using SigmaPlot software. Combined drug effects were analyzed by calculating the combination index (CI) with CalcuSyn software (Biosoft, Cambridge, UK). CI values of < 1, 1, and > 1 indicate synergistic, additive, and antagonistic effects, respectively.
8. Others
Further information on cell viability assay, colony-forming assay, western blot analysis, immunoprecipitation, cell cycle analysis, Annexin V/propidium iodide (PI) apoptosis assay, comet assay, transwell migration assay, and immunohistochemistry can be found in supplementary methods.
9. Ethical statement
Animal experiments were performed at the Biomedical Center for Animal Resource Development of Seoul National University (Seoul, Korea) according to institutional guidelines, and prior approval of the study protocol was obtained from the Institutional Animal Care and Use Committee.
Results
1. WEE1 inhibition inhibits the proliferation of PC cells
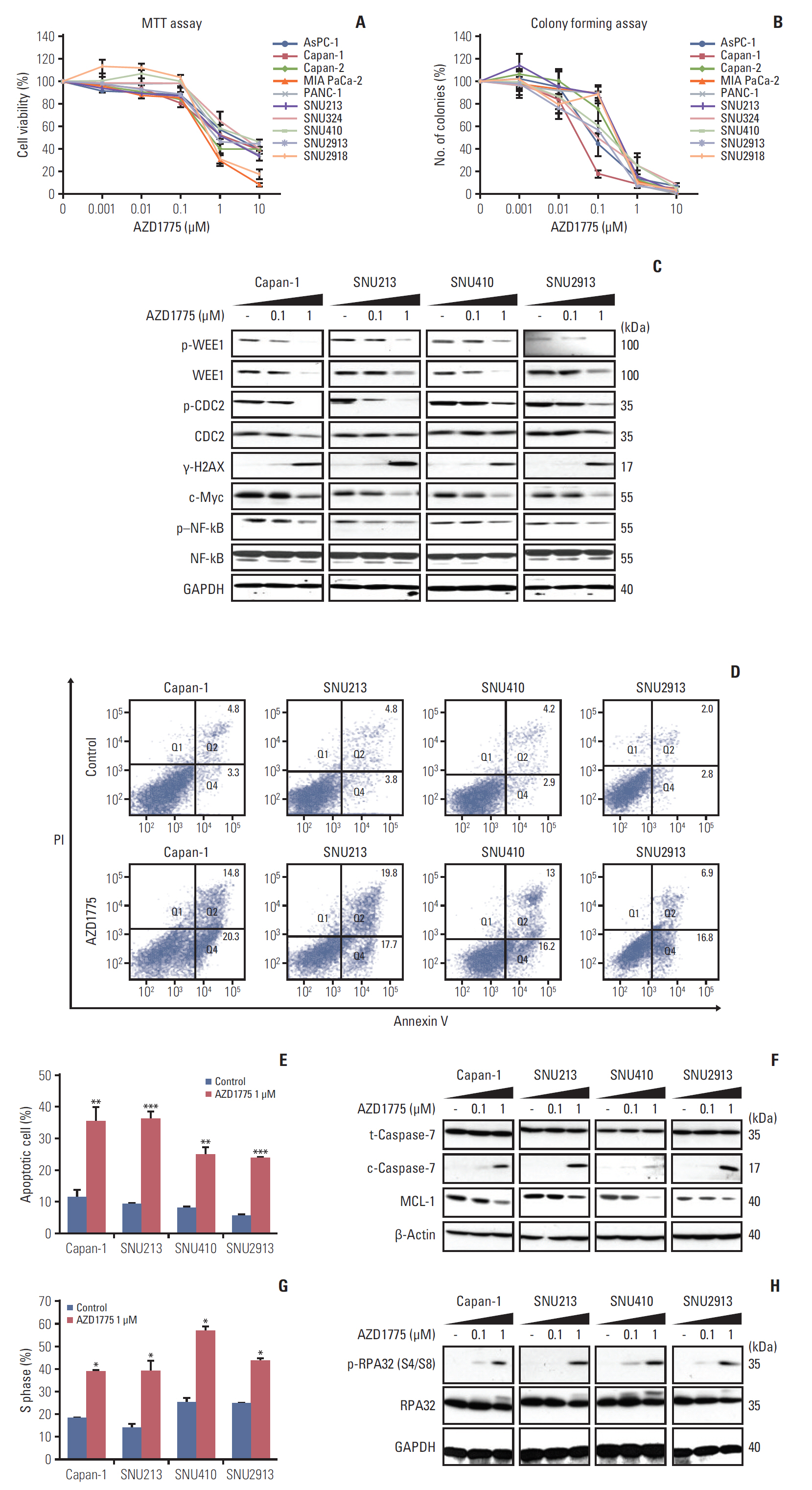
To evaluate the anti-tumor effects of WEE1 inhibition in PC, we used the MTT assay to assess the proliferation of 10 human PC cell lines in the presence of AZD1775 for 72 hours. As shown in Fig. 1A, AZD1775 inhibited the proliferation of all PC cell lines in a dose-dependent manner. The concentrations causing 50% inhibition (IC50) ranged from 0.5 to 2.1 μM (S1 Table). To verify these data, we examined the ability of the PC cell lines to form colonies after 10 days of incubation with AZD1775. This analysis also indicated a profound suppression of colony formation in all PC cell lines (Fig. 1B), and the low IC50 values for colony formation (0.03-0.36 μM) confirmed the sensitivity of human PC cells to WEE1 inhibition (S1 Table). Because all 10 of the PC cell lines showed comparable inhibition by AZD1775, we randomly selected four cell lines (Capan-1, SNU213, SNU410, and SNU2913) for the following experiments.
To determine whether AZD1775 blocked signaling in the DDR pathway, we performed western blot analysis of the expression and activation (phosphorylation) of a number of molecules involved in DDR signaling downstream of WEE1. For these experiments, PC cells were incubated with or without AZD1775 for 24 hours before analysis by western blotting. As shown in Fig. 1C, we confirmed that AZD1775 dose-dependently reduced the expression of total and phosphorylated WEE1 (p-WEE1) in all PC cells tested. In addition, AZD1775 also decreased the expression of phosphorylated cell division cycle protein 2, c-Myc, and phosphorylated nuclear factor kappa-light-chain-enhancer of activated B cells (p–NF-kB), and upregulated the expression of γ-H2AX compared with control (Fig. 1C). These results suggest that AZD1775 monotherapy has anti-proliferative activity in PC cells.
2. WEE1 inhibition induces S-phase arrest and apoptosis in PC cells
Since WEE1 inhibition reduced PC cell proliferation, we next determined whether AZD1775 induced apoptosis. For this, the cells were incubated with or without AZD1775 for 48 hours and apoptosis was examined by flow cytometry of Annexin V/PI–stained cells or by western blot analysis of an apoptosis regulator, MCL-1, and an effector, cleaved caspase 7. We found that AZD1775 treatment significantly increased the proportion of apoptotic cells compared with control cells (Fig. 1D and E) and concomitantly diminished the expression of MCL-1 and elevated the expression of cleaved caspase-7 (Fig. 1F). Since WEE1 functions as a regulator of cell cycle progression, we also assessed the proportion of cells in the cell cycle phases by flow cytometric analysis of PI-stained cells. As shown in Fig. 1G, a significantly greater proportion of cells treated with AZD1775 than control was arrested in S-phase. To verify this, we examined expression of phosphorylated replication protein A 32 (p-RPA32), an S-phase marker that binds to single-stranded DNA. Indeed, AZD1775 treatment for 24 hours resulted in increased p-RPA32 at serine 4 and 8, indicating that WEE1 inhibition leads to replication stress (Fig. 1H).
3. Co-inhibition of WEE1 and ATM profoundly impairs activation of the HR pathway of DNA repair
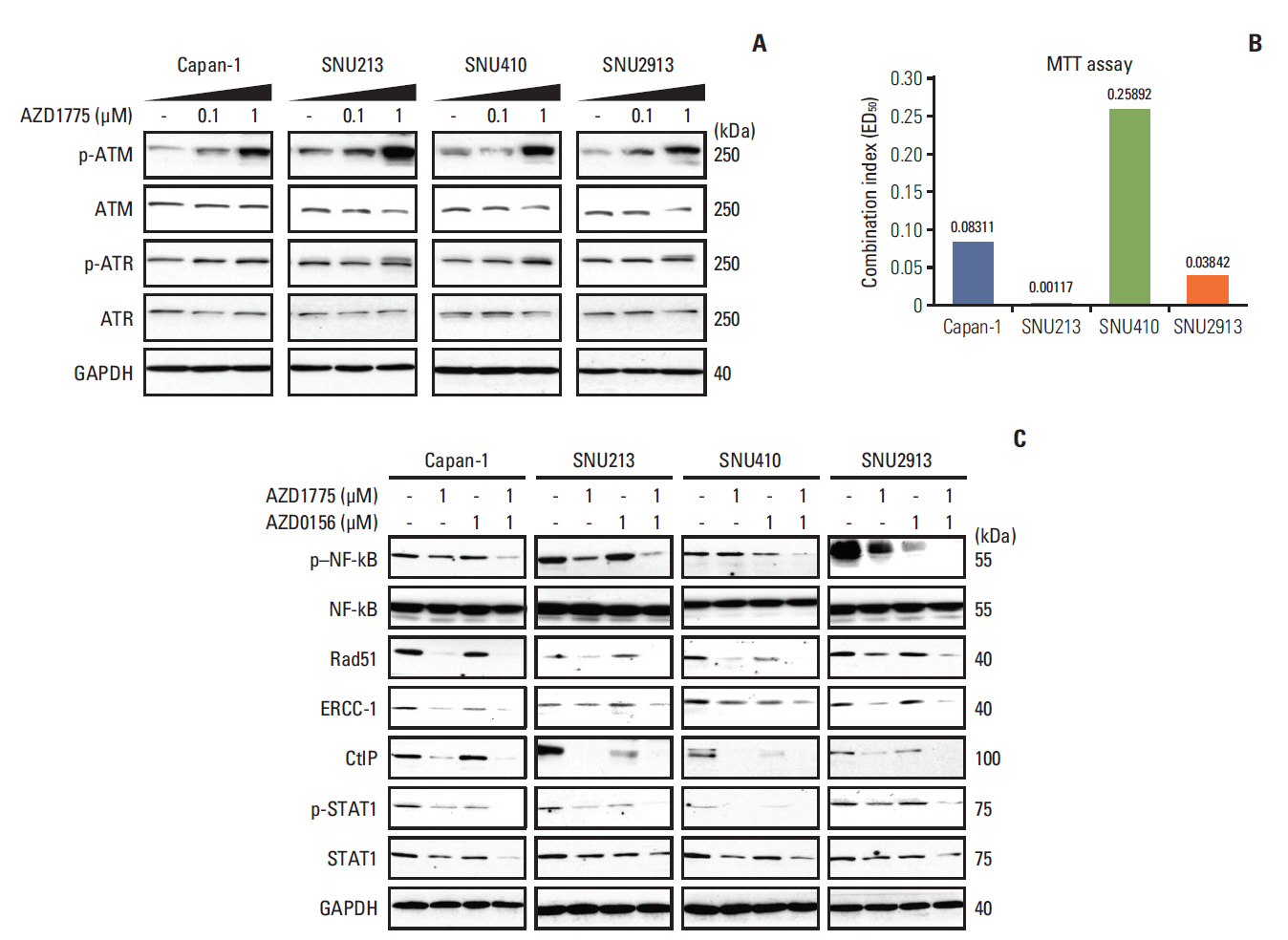
Having evaluated the effects of AZD1775 as a single agent, we asked whether WEE1 inhibition influences the activity of other core members of the DDR, such as ATM or ATR. Indeed, we found that phosphorylation of ATM and ATR was increased in cells treated for 24 hours with AZD1775 (Fig. 2A). Since AZD1775 had a greater effect on promoting phosphorylation of ATM than of ATR, we next examined the anti-proliferative effects of AZD1775 in combination with the ATM inhibitor AZD0156 using the MTT assay. As shown in Fig. 2B, the CI values of AZD0156 plus AZD1775 were less than 1 for all four PC cell lines, indicating that the drugs had a synergistic effect on proliferation.
To better understand the effects of combination AZD1775 and AZD0156 treatment, we examined the expression levels of several molecules required for DNA repair via HR, after treatment of cells for 72 hours with AZD0156 and/or AZD1775. We found that expression of p–NF-κB, Rad51, excision repair cross-complementing protein 1, C-terminal binding protein-interacting protein, and p-STAT1 were all downregulated by AZD1775 or AZD0156 when added alone, but a greater effect was observed in cells co-treated with both agents (Fig. 2C). These data suggest that co-inhibition of WEE1 and ATM strongly blocks the HR pathway of DNA repair.
4. Co-inhibition of WEE1 and ATM synergistically induces DNA damage
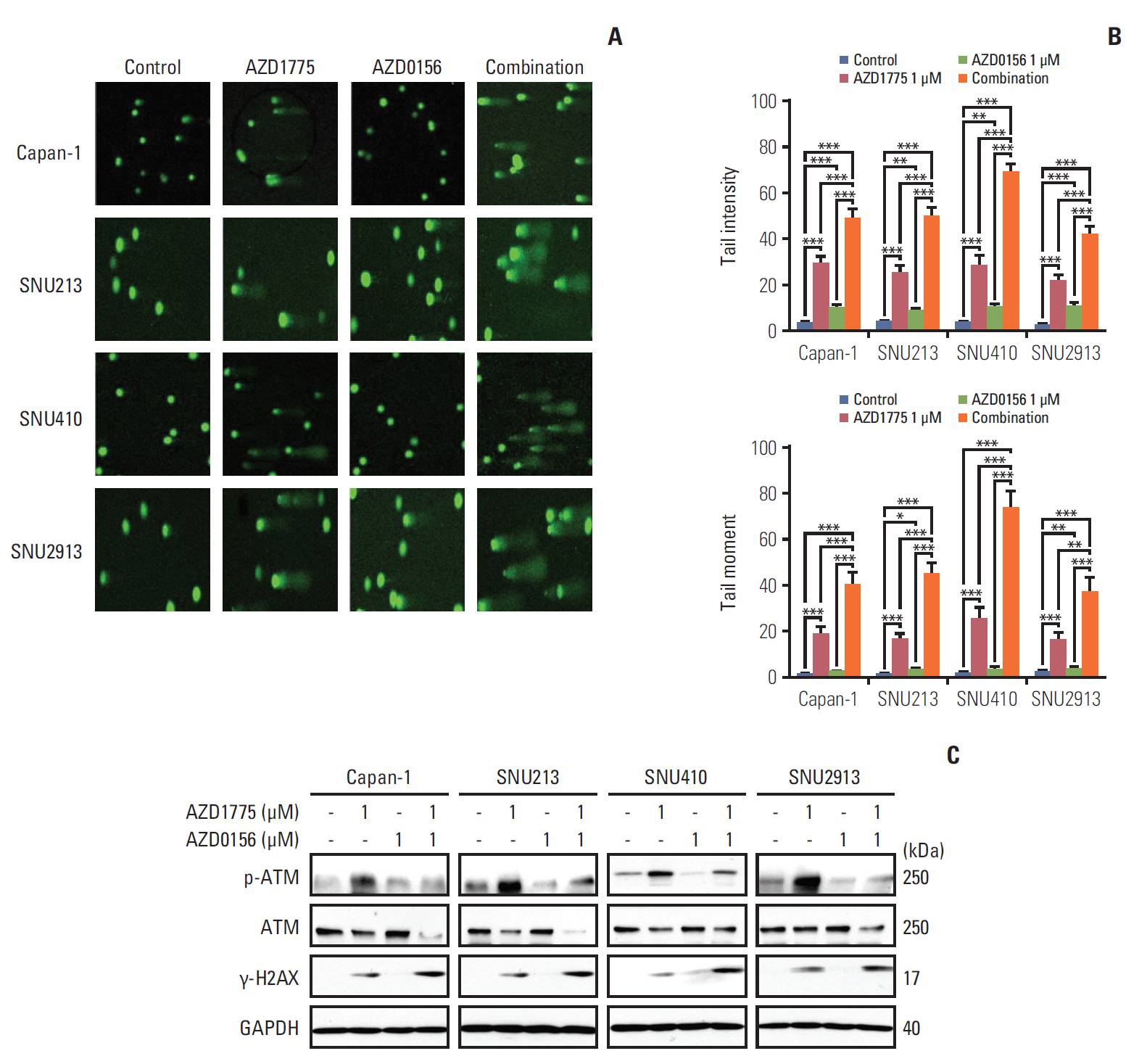
Having demonstrated that co-inhibition of WEE1 and ATM blocks DNA repair, we next asked whether these agents could induce DNA damage. We exposed the cells to AZD1775 and/or AZD0156 for 24 hours, and then monitored DNA fragmentation at the single-cell level using a comet assay. We found that AZD1775 or AZD0156 alone promoted DNA damage compared with control, but both agents in combination caused markedly increased fragmentation, as indicated by comet tail intensity and moment (Fig. 3A and B). Consistent with this, western blot analysis revealed upregulated expression of the DNA damage marker γ-H2AX, with a greater effect observed in cells subjected to combination AZD1775 and AZD0156 treatment compared with monotherapy (Fig. 3C).
5. Co-inhibition of WEE1 and ATM efficiently suppresses PC cell migration
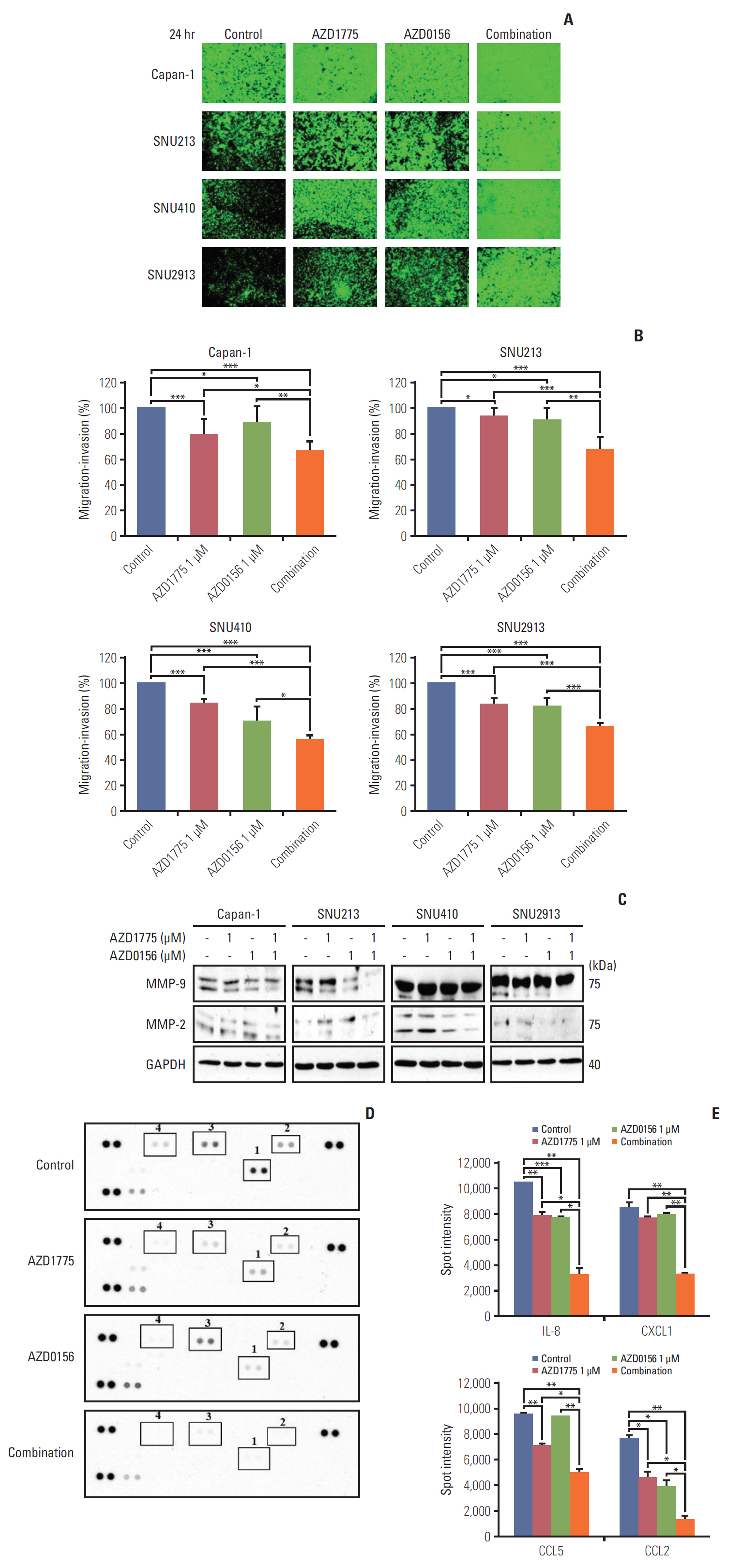
To determine the consequences of WEE1 and ATM inhibition on PC cell function, we examined cell migration and invasion, which play well-characterized roles in cancer progression. Importantly, very few studies have evaluated the effects of DDR-targeted agents on tumor cell migration. In this study, Transwell migration assay was employed. Interestingly, we found that cell migration and invasion of all four PC cell lines were significantly suppressed by treatment with 1 μM AZD1775 or AZD0156 alone. However, the combination of both drugs resulted in efficiently inhibition of cell migration (Fig. 4A and B). Thus, WEE1 and ATM appear to have previously unrecognized functions in promoting PC cell migration.
The matrix metallopeptidases 9 and 2 (MMP-9 and MMP-2) are known to be crucial for cancer metastasis and invasion [17,18]. Therefore, we asked whether the effects of WEE1 and/or ATM inhibition on PC cell migration were mediated via these enzymes. Western blot analysis revealed that treatment with AZD1775 or AZD0156 for 24 hours slightly decreased the expression of the smaller, active form of MMP-9 compared with control cells, and this effect was augmented by co-treatment with both agents (Fig. 4C). In contrast, a reduction in active MMP-2 expression was only observed in combination-treated Capan-1 and SNU410 cells (Fig. 4C).
Next, we investigated the effects of DDR targeting on chemokine expression in Capan-1 cells using a human cytokine/chemokine array. We found that the chemokines interleukin 8 (IL-8), CXCL1, CCL5, and CCL2 were significantly downregulated by AZD1775 treatment, combination therapy again showed an enhanced inhibitory effect on all four chemokines (Fig. 4D and E). Interestingly, previous work showed that signaling via the receptors for these chemokines (CXCR2, CCR5, and CCR2), facilitate the release of MMP-9, which contributes to enhanced angiogenesis and tumor metastasis [19-21]. Collectively, our results suggest that AZD1775 and AZD0156 act synergistically to suppress PC cell migration by reducing chemokine expression and MMP-9 release.
6. Co-inhibition of WEE1 and ATM downregulates PD-L1 expression in PC cells
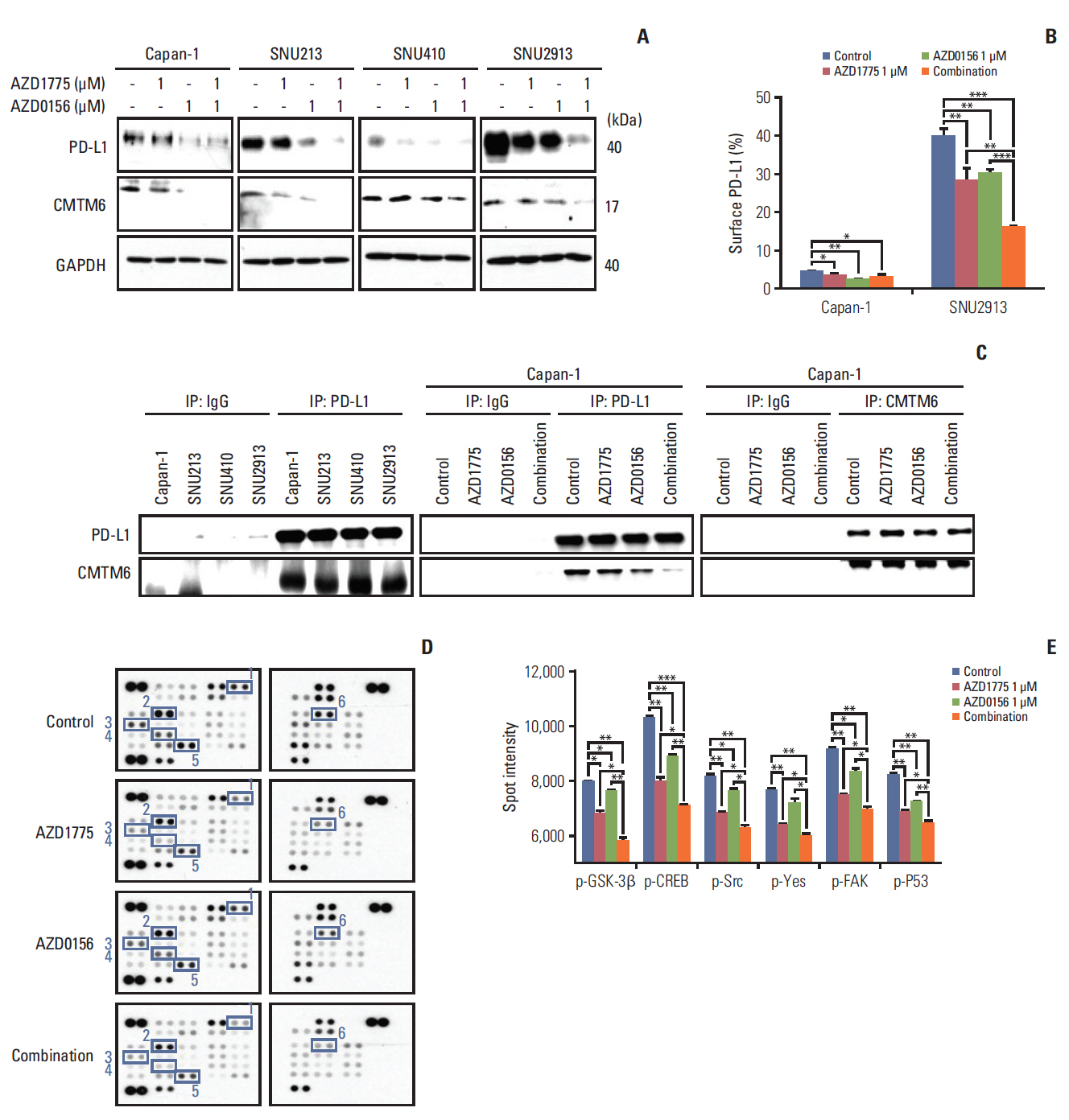
Next, given that the pivotal status of PD-L1 expression in cancer cell or cancer microenvironment [22], we explored the potential interaction between the DDR and the anti-tumor immune response by examining the effects of DDR-targeted agents on total and cell surface expression of PD-L1 in PC cells. Western blot analysis revealed that total cellular PD-L1 expression was decreased by AZD1775 or AZD0156 treatment alone, but combination treatment was even more effective (Fig. 5A). This pattern was also observed when cell surface PD-L1 expression was examined by flow cytometry, particularly in SNU2913 cells, which express high PD-L1 levels (Fig. 5B).
To understand in more detail, the effects of WEE1 and ATM inhibition on PD-L1 expression, we investigated potential mechanisms that regulate PD-L1: CMTM6, which is thought to prevent PD-L1 degradation by lysosomes [12], and GSK3β, which has recently been identified as a novel regulator of PD-L1 expression [23]. Western blot analysis of CMTM6 expression showed a marked decrease in the protein levels after treatment for 72 hours with AZD1775 and AZD0156, either alone or in combination (Fig. 5A). To probe CMTM6-mediated regulation of PD-L1 further, we asked whether this molecule is associated intracellularly by immunoprecipitating with anti–PD-L1 and probing for the presence of CMTM6 in the immunoprecipitates. We found that CMTM6 was present in western blots of anti–PD-L1 immunoprecipitates, but not control IgG immunoprecipitates, of all four PC cell lines (Fig. 5C). Notably, immunoprecipitation (IP) with anti– PD-L1 antibody showed the CMTM6–PD-L1 binding was decreased upon WEE1 or ATM inhibition, with the greatest effects observed upon dual inhibition. However, IP of CMTM6 followed by western blotting of PD-L1 did not show a comparable decrease in CMTM6–PD-L1 binding, even in cells co-treated with both AZD0156 and AZD1775 (Fig. 5C). It is possible that the reduction of CMTM6 by AZD1775 and AZD0156 led to decrease of CMTM6-PD-L1 bound form, which ultimately increase the PD-L1 degradation by lysosome supported by previous report [12].
To determine whether PD-L1 expression might be affected by the regulator GSK-3β in PC cells [23], we examined changes in phosphorylated kinase expression in AZD1775 and/or AZD0156-treated cells using a human phospho-kinase array. We observed that while AZD1775 and AZD0156 both downregulated p–GSK-3β expression, the combination treatment was more effective than either agent alone (Fig. 5D and E). This indicated that PD-L1 expression not only controlled by CMTM6 but also influenced by GSK-3β activity to a certain extent. Moreover, we found the same inhibitory effects on several other phospho-kinases, including phosphorylated cAMP response element-binding protein (p-CREB), p-Src, phosphorylated focal adhesion kinase (p-FAK), p-Yes, and p-p53 (Fig. 5D and E). Taken together, these results indicate that dual blockade of WEE1 and ATM may reduce PD-L1 expression by downregulating the expression of CMTM6, and inactive GSK-3β.
7. Co-inhibition of WEE1 and ATM augmented anti-tumor growth in Capan-1-xenograft model
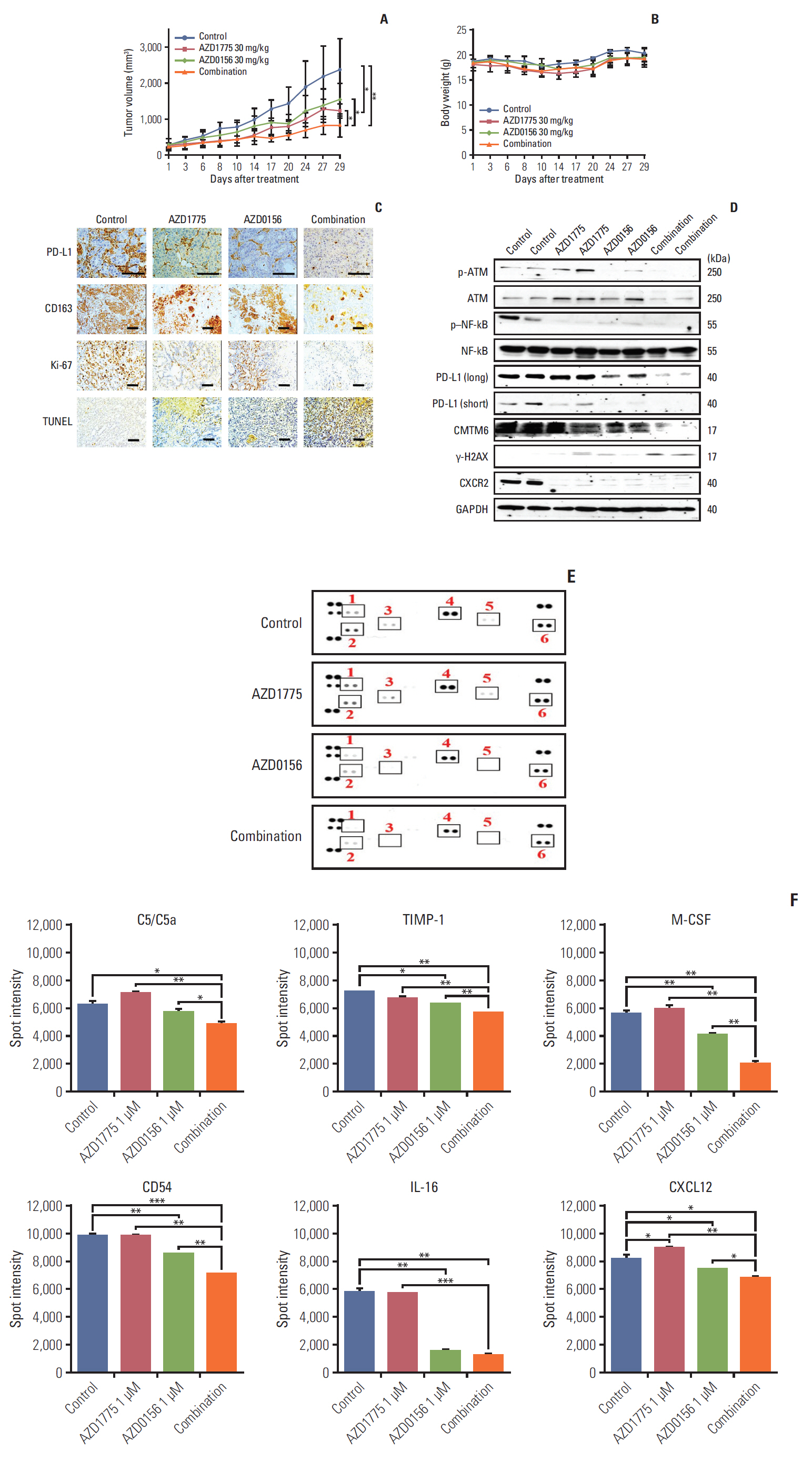
To confirm the anti-tumor effect of AZD1775 and AZD0156 in vivo, Capan-1 xenograft model was established. Despite high PD-L1 expression was detected in vitro in SNU2913 cells, since Capan-1 was more tumorigenic than SNU2913, we selected Capan-1 xenograft models to conduct the following experiments. Consistent with the in vitro observations, we found that single-agent treatment with AZD1775 or AZD0156 significantly decreased tumor growth, but the combination treatment was markedly more effective than either agent alone (p < 0.05) (Fig. 6A). We observed no overt evidence of drug toxicity, as reflected by a lack of significant change in body weight (Fig. 6B).
Next, we excised the tumors and performed immunohistochemical (IHC) staining of various markers. As we had observed in vitro, PD-L1 expression was decreased by AZD1775 or AZD0156 monotherapy and decreased by combination treatment (Fig. 6C). Moreover, CD163, an M2-type macrophage marker, was expressed at high levels in tumors from control mice but at lower levels in tumors from AZD1775- and/or AZD0156-treated mice (Fig. 6C). Combination treatment with both drugs also potently inhibited staining of the proliferation marker Ki-67 and increased terminal deoxynucleotidyl transferase dUTP nick end labeling staining, which detects fragmented DNA (Fig. 6C). In parallel, we performed western blot analysis to confirm our in vitro findings in the isolated tumors. In line with the in vitro results, we confirmed that AZD1775 treatment upregulated p-ATM while combination treatment with AZD-1775 and the ATM inhibitor reversed this. Moreover, the expression of p–NF-κB and CXCR2 were profoundly blocked by AZD1775 and/or AZD0156 treatment, and AZD1775 plus AZD0156 enhanced γ-H2AX accumulation. Furthermore, we validated the in vitro findings with PD-L1 and CMTM6 by confirming that both proteins were downregulated by single- or dual-agent treatment (Fig. 6D).
Finally, we assessed the effects of the DDR-targeting agents on immune cell activity by analyzing cytokine levels in the sera of tumor-bearing mice. Using a mouse cytokine array, we found that release of C5a, TIMP metallopeptidase inhibitor 1 (TIMP-1), macrophage colony-stimulating factor, CD54, IL-16, and CXCL12 were markedly reduced by AZD1775 and/or AZD0156 treatment, with larger effects observed in drug combination-treated mice (Fig. 6E and F). Collectively, these in vivo data confirm that dual targeting of the DDR pathway components WEE1 and ATM profoundly suppress tumor growth in vivo compared with blockade of either molecule alone.
Discussion
Although ATM is a tumor suppressor gene, both of WEE1 and ATM promote DNA repair to maintain cell survival during the progression of the cancers [24]. Inhibition of WEE1 and ATM activity are our opportunity to kill the cancers by facilitating DNA damage.
In this study, we demonstrate for the first time that targeting the DDR by inhibition of WEE1 has anti-tumor effects in PC and that co-inhibition of WEE1 and ATM amplifies these effects. We also identified crosstalk between the DDR pathway and immune system by demonstrating DDR-dependent regulation of PD-L1 expression in PC cells.
One of the main findings of this study is that dual inhibition of WEE1 and ATM has profound effects on PC cell migration. Only a few studies have been performed on the anti-migratory effects of DDR-acting agents. One report showed that AZD1775 inhibits the migration of gastric cancer cells, although the mechanism of action was not elucidated [25]. Another study suggested that ATM might promote cell migration by regulating IL-8 expression independently of its role in DNA double-strand break repair [26]. We found that WEE1 and ATM co-inhibition reduced the expression of MMP-9, IL-8, CXCL1, CCL2, and CCL5, which is consistent with an anti-migratory effect. This is the first demonstration of the involvement of WEE1 or ATM in MMP-9 or IL-8 regulation. Moreover, downregulation of CXCR2, which is the IL-8 and CXCL1 receptor, in PC tumors was confirmed in our in vivo experiments. Although CXCR2 inhibition was shown to profoundly suppress metastasis and augments anti–PD-1 therapy in PC [10], the current study is the first to examine CXCR2 expression in PC. Since CXCR2 and CCR2 are also expressed by M2 macrophages, we speculate that combination AZD1775 and AZD0156 treatment might impair M2 type polarization [27]. In addition, the involvement of Src and FAK in promoting cell migration and invasion is well known [28], and a recent report implicates a similar role for p-CREB [29]. Thus, our finding that expression of p-CREB, p-Src, p-Yes, and p-FAK are profoundly inhibited by WEE1 and ATM co-inhibition is consistent with a role for these molecules in PC cell migration. Prior to this study, there had been no reports of a relationship between Src/FAK/CREB and the DDR pathway in PC.
An interesting recent study reported that PARP inhibition upregulates PD-L1 expression in breast cancer via inactivation of GSK-3β [16]. We also found a similar increase in PD-L1 expression in PARP inhibitor (Olaparib)-treated PC cells (data not shown). In contrast, the present study showed here that targeting of the DDR by WEE1 and/or ATM inhibition reduced PD-L1 expression concomitantly with downregulation of p–GSK-3β Ser9 level, the inactive form of GSK3β. This mechanism might at least partly explain the reduction in PD-L1 induced by WEE1 and/or ATM inhibition. It is interesting to note that PARP, WEE1, and ATM have distinct effects on PD-L1 expression, despite the fact that they are all core members of the DDR signaling network. Thus, while PARP inhibition increases PD-L1 expression by enhancing GSK-3β inactivation, WEE1 and/or ATM inhibition does the opposite by decreasing the expression of inactive GSK-3β.
Among the mechanisms known to regulate PD-L1 expression, many act at the transcriptional level, including the JAKSTAT pathway, c-Myc, and NF-κB [15,30,31]. Consistent with this literature, we also observed a reduction in p-STAT-1 and p-NF-κB concomitant with PD-L1 downregulation in WEE1 and/or ATM inhibitor-treated cells. More recently, Burr and colleagues used CRISPR/Cas9 technology to screen approximately 20,000 genes in the human PC cell line BxPC-3, and they identified a novel protein, CMTM6, as a major regulator of PD-L1 expression [12]. Importantly, we observed that CMTM6 binds to PD-L1 in PC cells and that CMTM6 expression was reduced upon inhibition of WEE1 and/or ATM. As mentioned earlier, CMTM6 could rescue and recycle the PD-L1 from the lysosome to the cell surface. More recently, an interesting study revealed that HIP1R facilitates the delivery of PD-L1 to the lysosome [32]. Here, we speculated two potential mechanisms of the CMTM6 reduction by WEE1 or ATM. First of all, WEE1 or ATM might post-transcriptionally increase the HIP1R expression and compete for CMTM6 to bind with PD-L1. Secondly, CMTM6 might be transcriptionally downregulated by WEE1 or ATM, and these events further disrupt the interaction between PD-L1 and CMTM6. The mechanism by which DDR targeting blocks CMTM6 expression remains to be clarified.
As mentioned earlier, KRAS and TP53 are frequently mutated in PC. Recent work suggests that KRAS mutant cancer cells can enhance the recruitment of MDSCs and M2 macrophages to the tumor, thereby contributing to evasion of the immune response. M2 macrophages can also be recruited by cancer cells carrying mutant TP53 [9]. These macrophages promote tumor evasion through multiple mechanisms, including enhanced angiogenesis and tissue remodeling, recruitment of MDSCs and CAFs, and inhibition of T cell function and proliferation, thus enhancing metastasis and invasion [33]. Here, we evaluated M2 macrophage recruitment to the tumor in the mouse xenograft model by IHC staining of CD163, and we found that the high basal levels of tumor-associated CD163+ cells were markedly decreased by AZD1775 and/or AZD0156 treatment. This finding supports the possibility that combination therapy suppresses recruitment of M2 macrophages to the tumor, and it will be of interest to understand how other cells in the tumor microenvironment are affected by DDR-targeting agents.
We used high concentrations of WEE1 inhibitor (AZD1775-1 μM) and ATM inhibitor (AZD0156 1 μM) in current in vitro experiments considering the active pharmacology of compounds. Particularly, AZD0156 1 μM might have off-target effects.
Taken together, our results demonstrate that DDR-targeting agents such as WEE1 and ATM inhibitors have potent, synergistic anti-tumor effects in PC. We also identified crosstalk between the DDR and immune response in PC, since WEE1/ATM inhibition downregulates CXCR2, CD163, and PD-L1 expression, the latter by blocking expression of inactive p–GSK-3β and CMTM6. The findings in this study support further clinical development of DDR-targeting strategies for the treatment of PC.