Introduction
Malignant melanoma is uncommon but aggressive and is the most fatal type of skin cancer. This cancerous growth arises from a melanocyte and can spread to other parts of the body more rapidly than other cancers. Approximately, 87,110 new cases and 9,730 deaths from melanoma have been reported in the year 2017 in the United States [1]. Though melanoma constitutes less than 5% of skin cancers, it carries a high mortality rates. Melanoma risk factors include both intrinsic and extrinsic features. Phenotypes such as fair skin, blond hair, freckles, and blue eyes are commonly present in melanoma patients and are considered typical intrinsic risk factors [2]. In addition to phenotype, melanoma progression can be attributed to sun exposure (an extrinsic factor), family history, and genetic factors [3]. Furthermore, melanoma patients commonly present with mutations in genes that contribute to melanoma development [4,5]. In particular, the majority of melanoma patients possess driver oncogenic mutations in genes that encode proteins associated with mitogen-activated protein kinase/ERK and phosphoinositide 3-kinase/Akt (growth factor receptors) signaling pathways [6]. About 50% of melanoma cases hold BRAF mutations that generally substitute valine with glutamic acid in position 600 (V600E), and about 20%-30% of melanoma cases contain RAS mutations, which was the first identified oncogene relating to melanoma [7, 8]. Recent research on developing malignant melanoma therapies has focused on specific targeted therapies using BRAF and MEK inhibitors and introduction of immune checkpoint blockades such as anti–cytotoxic T-lymphocyte–associated protein 4, anti–programmed cell death protein 1, and anti–programmed death-ligand 1. As each therapy has its own limitations in response rate or duration, combined treatment of targeted inhibitors and immune checkpoint inhibitors has been suggested to treat malignant melanoma [9]. However, patients who were exposed to these types of therapy gained resistance to the treatments, which led researchers to seek an alternative method of therapy for melanoma.
Gene therapy is one potential candidate for alternative melanoma treatments. More specifically, gene-directed enzyme/prodrug therapy (GDEPT) has been studied as a prominent tool for treating cancers through molecular chemotherapy [10]. Unlike conventional chemotherapies, the GDEPT system minimizes the toxicity of drugs in normal tissues, and neural stem cell-directed enzyme/prodrug therapy (NDEPT), a suicide gene therapy, was developed to selectively target cancers while reducing the damages to normal tissues [11]. Suicide gene therapy makes use of the bystander effect of a suicide enzyme, which converts an inactive drug to an active drug and causes cell death in tumors [12]. Though various suicide gene systems exist, the cytosine deaminase (CD)/5-Fluorocytosine (5-FC) system was applied in this study. CD impedes DNA synthesis and enhances apoptosis in tumor cells by modifying the inactive drug 5-FC into its active metabolized by-product 5-fluorouracil (5-FU) [13].
In a similar fashion, the cytokine interferon-β (IFN-β), is able to promote cell cycle arrest in S-phase and apoptosis in tumor cells [14]. Notwithstanding the therapeutic effect of IFN-β at a high concentration in vitro, its use in vivo causes side effects and limits its therapeutic application in high doses [15]. We utilized human neural stem cells (hNSCs) HB1.F3 that were obtained from 15-week-old fetal telencephalon, and immortalization was performed using a retroviral vector encoding the v-myc oncogene. These hNSCs were transduced into two types: one expressing only cytosine deaminase (HB1.F3.CD) and the other expressing both CD and human IFN-β (HB1.F3.CD.IFN-β). The clonal HB1.F3.CD expressing only CD was generated by transfection of the Escherichia coli CD gene to immortalized hNSCs [16]. Neural stem cells are applicable as a therapeutic delivery vehicle for gene therapy because neural stem cells effectively migrate to the target tumor site by following chemoattractant and growth factors emitted by tumor cells [17]. It has been shown that many chemokines, growth factors and receptors mediate the migratory behavior of hNSCs in vitro due to the interaction of cytokine/receptor pairs such as stromal cell-derived factor 1 (SDF-1)/CXCR4, vascular endothelial growth factor (VEGF)/vascular endothelial growth factor receptor (VEGFR), SCF/c-kit, and MCP-1/CCR2 [18-20]. They can also be propagated for long periods. There have been several preclinical models demonstrating the therapeutic potential of engineered hNSCs, as they migrated to cancer cells selectively and hindered tumor cell growth both in vitro and in vivo for various cancers [10,21,22].
Though the treatments of malignant melanoma have become more advanced, they could not avoid side effects including damage to normal tissues and acquisition of resistance to the therapies. On the other hand, neural stem cellbased therapies have emerged as a feasible drug-delivery mechanism for various types of cancers due to their tumortropic behavior. The purpose of this study was to examine whether hNSCs expressing CD and/or IFN-β could migrate to malignant melanoma and thereby serve as a potential therapy vector for melanoma by co-culturing them both in vitro and in vivo with a malignant melanoma cell line (A375SM) in the presence of the prodrug 5-FC. We characterized the therapeutic effect of engineered hNSCs on melanoma through their tumor-tropic behavior using xenograft mouse models. Our results elucidate the suitability of neural stem cellbased therapy for treating malignant melanoma and demonstrate its promise as a tool for minimizing side effects through the tumor-tropism of neural stem cells.
Materials and Methods
1. Cell culture and media
A malignant melanoma cell line, A375SM, was bought from Korean Cell Line Bank (KCLB, Seoul, Korea). The three engineered hNSC lines (HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β) used in this study were provided by Dr. Seung U. Kim (University of British Columbia, Vancouver, BC, Canada). A primary human fibroblast cell line (hFB) was also used for a modified Boyden chamber migration assay as a control. Every cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone Laboratories, Logan, UT) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Hyclone Laboratories), 10 mM HEPES (Invitrogen Life Technologies, Carlsbad, CA), 10 U/mL penicillin and 100 μg/mL streptomycin (Cellgro Mediatech, Manassas, VA), and anti-mycoplasmal agents (Invivogene, San Diego, CA) at 37℃ in 5% CO2 and 95% air in a humidified cell incubator. Zero point zero five percent trypsin/0.02% EDTA (PAA Laboratories, Dartmouth, MA) was used to trypsinize the cells.
2. mRNA extraction and reverse transcriptase polymerase chain reaction
mRNA was extracted from melanoma cells and hNSCs using TriZol Reagent (Invitrogen Life Technologies). As per the manufacturer’s protocols, 1 μg of total mRNA, quantified with a spectrophotometer (Optizen, Mecasysm, Daejeon, Korea), was reverse transcribed into cDNA by applying murine leukemia virus reverse transcriptase (MMLV-RT, iNtRON Biotechnology, Seongnam, Korea), 200 pM nonamer random primer (TaKaRa Bio, Shiga, Japan), RNase inhibitor (iNtRON Biotechnology), 10 pM dNTP (Bioneer, Daejeon, Korea), and 5× RT buffer. Polymerase chain reaction (PCR) was conducted to confirm the expression of the bacterial CD gene and human interferon-β gene in hNSCs using their respective primers. PCR was also performed using the cDNA of melanoma cells and hNSCs to amplify the chemoattractant ligands and receptors including SDF-1, CXCR4, VEGF, VEGFR2, urokinase-type plasminogen activator and its receptor with sense and antisense primers based on the published sequences of the genes. As a positive control, the human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used. The PCR reactions were carried out for 30 cycles with denaturation for 30 seconds at 95℃, annealing for 30 seconds at 58℃, and extension for 30 seconds at 72℃. PCR reaction products (11 μL) were analyzed on a 1.5% agarose gel prestained with ethidium bromide (Sigma-Aldrich, St. Louis, MO) and NEOgreen (NEO Science, Suwon, Korea). The gels were scanned by a Gel Doc 200 apparatus (Bio-Rad Laboratories, Hercules, CA).
3. Cell viability assay
To evaluate the effect of 5-FU (Sigma-Aldrich) and 5-FC (Sigma-Aldrich) on melanoma cells (A375SM), 4,000 melanoma cells were seeded per well in 96-well plates (SPL Life Science, Seoul, Korea) and cultured in phenol free DMEM containing 5% charcoal-dextran treated FBS, 1% penicillin and streptomycin, and 1% HEPES at 37℃ in a humidified atmosphere of 95% and 5% CO2. After 24 hours of incubation, every well was treated with 5-FU or 5-FC diluted with phosphate buffered saline (PBS; 100, 200, 300, 400, and 500 μg/mL) for 3 days. To verify the therapeutic effect of engineered hNSCs (HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β), melanoma cells were seeded in the same media (4,000 cells/well) on day 1. Three types of engineered neural stem cells (16,000 cells/well) were added to the cancer cells on the next day. On the third day, cells were treated with a range of 5-FC concentrations diluted with PBS (100, 200, 300, 400, and 500 μg/mL) for 3 days. After 3 days of the treatment with 5-FU and 5-FC, an MTT (3-(4-,5-dimethylthiazol-2-yl)-2,5-dyphenyl tetrazolium bromide) assay was conducted to measure melanoma cell viability. MTT (Sigma-Aldrich) stock solution (10 μL; 5 mg/mL) was added to every well and incubated for 3 hours at 37℃. After removing supernatants, precipitated formazan crystals were dissolved in 100 μL of dimethylsulfoxide (99%, Junsei Chemical, Tokyo, Japan). Using an enzyme-linked immunosorbent assay (ELISA) plate reader (VERSA man, Molecular Devices, Sunnyvale, CA) at 540 nm, absorbance of each well was measured. Every step was performed in triplicate.
4. In vitro migration assay
To observe the ability of hNSCs to migrate to melanoma and normal human lung cells, both A375SM and L132 (1×105 cells/well) were plated in a 24-well plate containing 5% CD-FBS phenol free DMEM and incubated for 24 hours. The bottom surface of the transwell with 8.0-μm pores (BD Biosciences, Dickinson, Bedford, MA) was coated with fibronectin (25 μg/mL, Sigma-Aldrich) and placed in 24-well plates and engineered hNSCs (1×105 cells/well) were seeded in the upper compartment of the transwell on the next day. After 24 hours of incubation, all lower chambers were washed with PBS, migrated hNSCs were fixed using 3.7% formaldehyde solution (Sigma-Aldrich) for 15 minutes, and the cell underwent permeabilization using 10% coldmethanol (Sigma-Aldrich) for 15 minutes. Crystal violet was used to stain the surface of the transwell for 15 minutes and all transwells were rinsed thoroughly with PBS. Crystal violet stained hNSCs were visualized by fluorescence microscopy (IX-73 Inverted Microscopy, Olympus, Tokyo, Japan) and were counted with Cell Sense Dimension.
5. Xenograft model of metastatic melanoma
All experiments containing animals were approved by the Animal Care Committee of Chungbuk National University. Four-week-old athymic female nude mice were purchased from KOATECH (Pyeongtaek, Korea). All mice were kept in a room for a week to adapt to their environment (at 24℃, 40%-50% humidity and 12-hour light-dark cycle) before the experiments. To create melanoma xenograft models, A37-5SM cells (2×106) and matrigel (BD Biosciences) were mixed in a 1:1 ratio to a total volume of 200 L and subcutaneous injection was performed into the back of the mice as previously shown [23]. The tumor volume of every mouse was measured every other day with a caliper using the formula length×width×height×0.5236 (mm3).
6. Therapeutic efficacy of HB1.F3.CD and HB1.F3.CD.IFN-β hNSCs in vivo model
To evaluate the therapeutic effect of HB1.F3.CD and HB1.F3.CD.IFN-β in a melanoma xenograft model, the mice were split into four groups: group 1 was treated with the vehicle (5-FC; 500 mg/kg/day) as a control and groups 2, 3, and 4 were treated with the engineered hNSCs: HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β, respectively. When the tumor volume of each mouse reached between 500 mm3 and 600 mm3, CM-DiI pre-labeled HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β cells (8×106 cells/mouse) were weekly injected subcutaneously near the tumor site with matrigel at a 1:1 ratio for 3 weeks. Two days after the injection of engineered hNSCs, 5-FC (500 mg/kg/day) was intraperitoneally injected every day for 3 days. The animal study was conducted for 21 days after the injection of engineered hNSCs. To examine the therapeutic efficacy of engineered hNSCs, the tumor volume was measured for the last time before the mouse was euthanized 24 hours after the last injection of 5-FC. All tumor masses were dissected for molecular analysis. The tumor volumes were quantified by Graphpad Prism (v5.0, GraphPad Software, San Diego, CA).
7. Western blot assay
Proteins used for western blot were harvested using RIPA buffer containing 50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1% Triton X-100 (Sigma-Aldrich); 0.1% sodium dodecyl sulfate; and 0.5% deoxycholic acid (Sigma-Aldrich) mixed with protease and phosphatase inhibiter. Extracted proteins were incubated at 4℃ overnight then centrifuged at 14,000 rpm for an hour. Following centrifugation, protein concentration was decided using a mixture of bicinchoninic acid (Sigma-Aldrich) and copper (II) sulfate (Sigma-Aldrich). Fifty micrograms of the proteins were distributed on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis then separated proteins were transferred to polyvinylidene fluoride membrane (BioRad Laboratories Inc.). The membrane was then incubated at room temperature with 5% blocking buffer (BioRad Laboratories Inc.) to prevent non-specific binding with the primary antibody. Subsequently, the membrane was incubated with monoclonal primary antibody, mouse monoclonal anti-BAX (1:500 dilution, Cell Signaling Technology Inc.), anti-Slug (1:1,000 dilution, Abcam, Cambridge, UK), anti-GAPDH (1:3,000 dilution, Abcam), rabbit monoclonal anti–Bcl-xL (1:1,000 dilution, Cell Signaling Technology Inc.), anti–E-cadherin (1:500 dilution, Abcam), and anti–cathepsin-B (1:200 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4℃. The primary antibody binding was visualized by incubating with a secondary antibody, anti-rabbit IgG or anti-mouse IgG (1:3,000 dilution, Thermo Scientific Corp., Waltham, MA), which was conjugated with horse radish peroxidase for at least 1 hour at room temperature. Proteins were detected using a West-Q Chemiluminescent Substrate Plus kit (GenDEPOT, Barker, TX). Each protein was quantified using the densities of bands on a membrane utilizing CSAnalyzer4. Experiments were performed a minimum of 3 times and expression levels of all the above proteins were normalized with GAPDH protein.
8. 5-FU treatment
To determine 5-FU (Sigma-Aldrich) effects in neural stem cell therapy, melanoma cells were treated with different concentrations of 5-FU (1 and 5 μg/mL) for 0, 1, 3, or 6 hours; and proteins were extracted after 5-FU treatments. To evaluate the expression of the apoptosis-related genes BAX and Bcl-xL, A375SM cells were exposed to 1 and 5 μg/mL of 5-FU for 0, 1, 3, and 6 hours.
9. HB1.F3.CD and HB1.F3.CD.IFN-β conditioned media
The engineered hNSC lines HB1.F3.CD and HB1.F3.CD.IFN-β were cultured in DMEM with 10% heat-inactivated FBS, 1% HEPES, and penicillin/streptomycin in a T75 flask until cells were 70% full at 37℃ in 5% CO2. Engineered neural stem cell lines were then incubated with new media. Media were filtered using a 0.22-mm syringe filter after 2 days of incubation then used for melanoma cultures for 0, 24, 48, and 72 hours.
10. Immunohistochemical assay
Excised tumors from mice were fixed in 10% formalin solution (OCI, Seoul, Korea). Fixed tumors were cut into 5-mm thick slices, dehydrated, and embedded in paraffin. Paraffin blocks were sectioned into 5-μm slides. Slides were placed in a chamber containing 0.01 M citrate buffer (pH 6.0) and microwaved for 10 minutes to retrieve tissue antigens. Following antigen retrieval, the slides were incubated with 0.3% H2O2, blocking buffer to reduce background staining, and the monoclonal primary antibody proliferating cell nuclear antigen (PCNA; 1:100 dilution, Abcam) overnight at 4℃. To evaluate the expression of primary antibody PCNA, biotinylated mouse IgG (1:1,000 dilution, Vector Laboratories, Burlingame, CA) was applied as a secondary antibody. For the digital photography of PCNA expression of each group, an IX-73 inverted microscope was used.
11. Fluorescent DAPI analysis
The distribution of hNSCs can be inspected in a tumor mass by pre-staining with CM-DiI before hNSC injection. Dehydrated tissue slides were fixed in 10% formalin, washed with PBS twice, and DAPI solution was applied for 10 minutes at room temperature. DAPI stained slides were mounted with coverslips before fluorescent microscopy (IX-73 inverted microscopy) was used to observe them.
12. Data analysis
in vitro and in vivo data were statistically analyzed using GraphPad Prism (v5.0, GraphPad Software). In vitro experiments were completed a minimum of three times to ensure consistent results. In vitro data were shown as the mean±standard deviation and in vivo data were presented as the mean±standard error of the mean. To examine the significance of in vitro and in vivo data, one-way analysis of variance (ANOVA) followed by post hoc Dunnett’s or Tukey’s test were used. p-values of < 0.05 were regarded as statistically significant.
Results
1. Engineered Escherichia coli CD and human IFN-β genes were identified in engineered neural stem cell lines
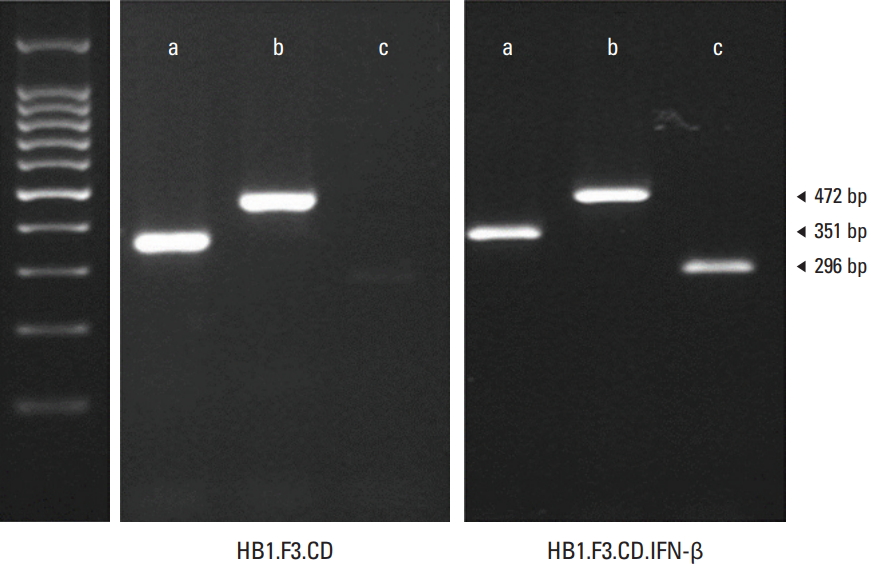
Reverse transcriptase–polymerase chain reaction (RT-PCR) was carried out for verifying the expression of the inserted genes: Escherichia coli CD and IFN-β. The expression of the CD gene (472 bp) was detected in the HB1.F3.CD cell line and the expression of both CD and human IFN-β gene (296 bp) were detected at in the HB1.F3.CD.IFN-β cell line as shown in Fig. 1. The GAPDH gene was used as a standard for RT-PCR and detected at 351 bp in both the HB1.F3.CD cell line and HB1.F3.CD.IFN-β cell line.
2. hNSCs were able to migrate toward melanoma
A modified Boyden chamber assay was used to observe the migration capability of engineered hNSCs toward melanoma cells. Both A375SM cells and normal human lung cells were seeded (1×105 cells/mL) in 24-well plates for 24 hours then the same number of hNSCs (HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β) were seeded on the upper compartment of the chambers for another 24 hours. As shown in Fig. 2A, hNSCs that migrated were stained with crystal violet and viewed using fluorescent microscopy. They were counted and the ratio of hNSCs that selectively migrated towards melanoma cells to those that migrated toward normal human cells was measured using fluorescent microscopy. hNSCs migrated to melanoma cells about 4 times more often than they migrated to normal human cells (Fig. 2B), which implied that hNSCs were tumor-tropic. The migration of neural stem cells could be explained through the interaction between chemoattractant ligands and their receptors with tumors. Expression of chemoattractant ligands and the parallel receptors in melanoma cells or hNSCs was confirmed by their RNA levels using RT-PCR. Melanoma cells expressed chemoattractant ligands, e.g., SDF-1 (263 bp) and VEGF (377 bp), and hNSCs expressed the corresponding chemoattractant factors, CXCR4 (298 bp) and VEGFR (143 bp) (Fig. 2C).
3. Expression of CD and IFN-β genes in hNSCs showed anti-proliferative effects on melanoma in the presence of 5-FC in vitro
To verify the cytotoxicity of 5-FC and 5-FU on melanoma cells, A375SM cells were seeded and 5-FC or 5-FU were applied at increasing concentrations (100 to 500 μg/mL) on successive days for 3 days. The cell proliferation was quantified using an MTT assay. Proliferation of 5-FC groups was reduced slightly and there was no apparent concentration-dependent difference. However, cell proliferation of 5-FU groups decreased drastically even at 100 μg/mL concentration (Fig. 3A). To demonstrate the therapeutic efficacy of hNSCs on melanoma cells, A375SM cells were co-cultured with hNSCs in the presence of serial concentrations of 5-FC (100 to 500 μg/mL). The cell viability of A375SM was reduced noticeably when co-cultured with HB1.F3.CD and HB1.F3.CD.IFN-β with 5-FC treatment compared to the control group (HB1.F3). 5-FC hindered cell proliferation of A375SM cultured with HB1.F3.CD and HB1.F3.CD.IFN-β in a dose dependent manner (Fig. 3B). However, there was no synergistic effect of HB1.F3.CD.IFN-β on A375SM cells.
4. Tumor volume regressed after hNSC treatments in a xenograft mouse model
Xenograft mouse models were used to validate the antitumor effect of hNSCs through an in vivo study and were handled as shown in Fig. 4A. A375SM cells were subcutaneously injected on the back of female athymic mice to create melanoma xenograft models. The tumor volume of every mouse was measured using a digital caliper throughout the entire experimental period. NSC treatment was initiated when the tumor volume reached between 500 and 600 mm3 and hNSCs were injected nearby a tumor to give a practical approach. From the 14th day of hNSC treatment, the growth of tumor volume treated with HB1.F3.CD and HB1.F3.CD.IFN-β was significantly repressed compared to controls (vehicle or HB1.F3) and there was no synergistic effect of HB1.F3.CD.IFN-β, which corresponded to the result of the in vitro cell viability assay. Tumor volume of the HB1.F3 treated group or only 5-FC treated group gradually increased (Fig. 4B). The inhibitory effect presents in both HB1.F3.CD and HB1.F3.CD.IFN-β groups but not in the HB1.F3 group suggests that therapeutic genes engineered for expression in hNSCs prevented tumors from expanding in xenograft models. In addition, necrosis of the tumor mass started to occur on most of the mice that were treated with only 5-FC once the tumor volume reached more than 1,500 mm3.
5. Engineered hNSCs migrated towards cancer and inhibited proliferation of tumor mass
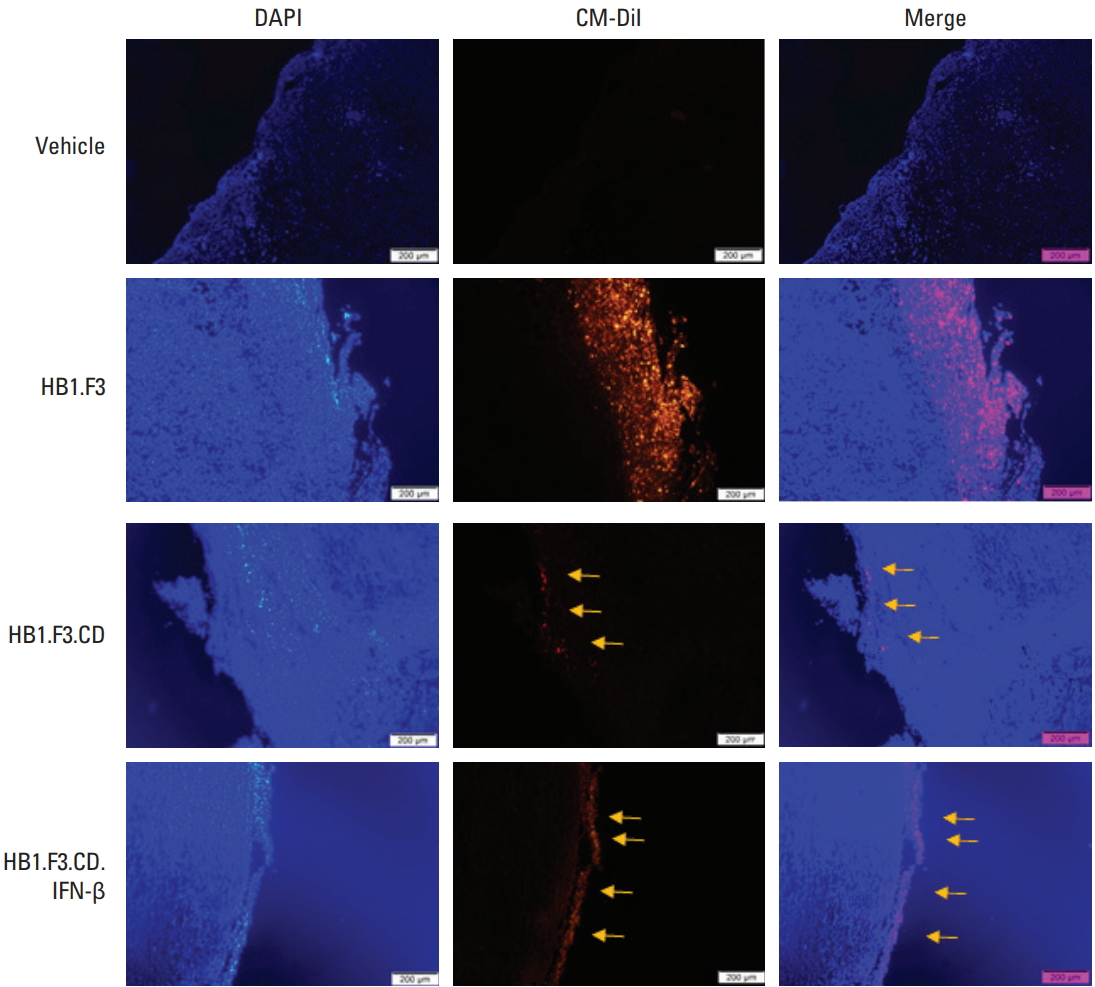
The migration of hNSCs toward tumors was observed through fluorescence spectroscopy. hNSCs were stained in red because of the CM-DiI cell tracker and the tumor mass was stained in blue with DAPI. The trace of CM-DiI prelabeled hNSCs on the surface and inside of the tumor in the HB1.F3, HB1.F3.CD and HB1.F3.CD.IFN-β groups (as shown with yellow arrows) verified that hNSCs migrated toward tumors via a chemoattractant ligand-receptor interaction in vivo (Fig. 5). Since there was no hNSC treatment in the vehicle group, there was no trace of the CM-DiI cell tracker. Furthermore, CM-DiI was more highly expressed in the HB1.F3 group compared to both the HB1.F3.CD and HB1.F3.CD.IFN-β group because the CD gene could be eliminated in the presence of 5-FC [24].
To learn if engineered hNSCs have an effect on tumor proliferation, paraffin-embedded melanoma was incubated with a primary antibody specific for PCNA, a proliferation marker of cancer. PCNA was highly expressed in both control groups, the vehicle group and the HB1.F3 group, but the expression in the HB1.F3.CD and HB1.F3.CD.IFN-β groups was manifestly low compare to the controls (Fig. 6). This result indicated that engineered neural stem cells converted 5-FC to 5-FU and impeded tumor proliferation in xenograft models.
6. 5-FU treatment altered the expression of apoptosisrelated protein levels in a melanoma cell line
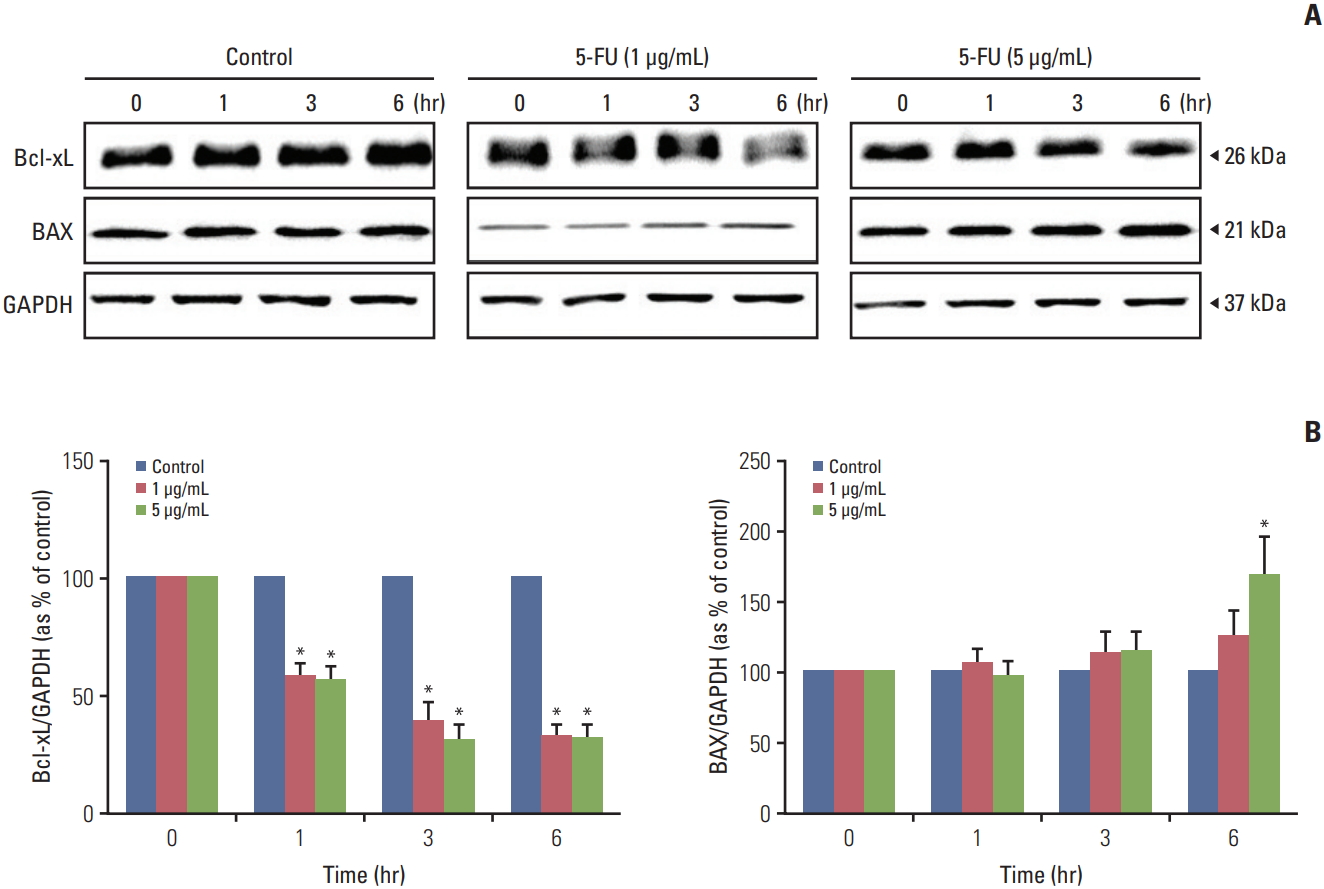
The alterations of apoptosis-related genes in melanoma cells after exposure to 5-FU were examined in this study using a western blot. Proteins from 5-FU (1 and 5 μg/mL) treated and untreated melanoma cells were extracted using RIPA buffer with a protease inhibitor after 0, 1, 3, and 6 hours. 5-FU treatment led to the decreased expression of the anti-apoptotic protein, Bcl-xL compared to the untreated protein in a time-dependent manner (Fig. 7). The data shown in Fig. 7 displayed a significant increase in the expression of the pro-apoptotic protein, BAX, for 5-FU (5 μg/mL) treated proteins after 6 hours compared to the proteins at 0 hour.
7. Human IFN-β from HB1.F3.CD.IFN-β cell line influenced melanoma in epithelial-mesenchymal transition and metastasis
To determine the efficacy of recombinant human IFN-β in the HB1.F3.CD.IFN-β cell line on melanoma, HB1.F3.CD.IFN-β was cultured in a T75 flask and filtered using a 0.22 mm syringe filter. We hypothesized that HB1.F3.CD.IFN-β cells secrete IFN-β while growing in the media and the secreted form of IFN-βwould have an effect on melanoma other than a synergistic effect. The filtered media was used for the IFN-β treatment on melanoma (the HB1.F3.CD cell line was used as a control and was handled in the same way as the HB1.F3.CD.IFN-β cell line). To evaluate the effect of the recombinant IFN-β on the epithelial-mesenchymal transition (EMT) and metastasis, IFN-β treatment on melanoma was performed for 0, 24, 48, and 72 hours. Proteins from IFN-β treated and untreated melanoma cells were harvested using RIPA buffer containing protease inhibitor. Among melanoma cells that were cultured with HB1.F3.CD.IFN-β conditioned media, the expression of the epithelial marker E-cadherin gradually increased in a time-dependent manner, and, at 72 hours, it was expressed twice as often as at 0 hours. On the other hand, the expression of the mesenchymal marker Slug and the metastasis marker cathepsin-B was diminished in a time-dependent manner (Fig. 8B). However, the melanoma cells that were cultured with HB1.F3.CD conditioned media had no significant changes in expressions of E-cadherin, Slug, and cathepsin-B after 72 hours as shown in Fig. 8A.
Discussion
Surgical excision of a tumor is a conventional treatment of melanoma, and radiation therapy using X-ray beams is often used after surgery to reduce local recurrence. While these conventional therapies are effective palliatives, they simultaneously damage other normal tissues and can fail to prevent melanoma from metastasizing. Once melanoma starts to metastasize to other organs, conventional therapies become more challenging to apply. The disease becomes unreachable if melanoma disseminates to the lymph nodes, lungs, brain, liver and bones, all of which are typical metastatic sites for melanoma. The development of molecular-targeted therapies (BRAF inhibition and MEK inhibition) increased survival rates for melanoma patients, but repeated exposure to the therapy resulted in patients acquiring a resistance to the treatment [25]. Hence, searching for an effective treatment for malignant melanoma is critical.
NDEPT is a plausible treatment for malignant melanoma due to its intrinsic migratory and tumor-tropic behaviour. Its properties are achieved through an interaction between chemoattractant and growth factors emitted by both tumor cells and neural stem cells and can be used to avoid damage to normal tissues. Neural stem cells used in this study were manipulated to express CD and/or human IFN-β. The CD/5-FC system operates as follows: CD deaminates the noncytotoxic drug 5-FC into the cytotoxic drug 5-FU and hinders tumors from growing by promoting apoptosis in tumor cells. A synergistic effect was expected to occur by combining the cytokine IFN-β with the CD/5-FC system. NDEPT exhibited an outstanding antitumor effect in preceding preclinical studies on several types of cancers [11,21]. The present study further suggests that engineered hNSCs can also be used as a competent delivery vector for selectively treating malignant melanoma.
Neural stem cells (HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN-β) were genetically engineered by retroviral transduction. The expressions of the inserted genes, CD and IFN-β, were confirmed through RT-PCR. The tumor-tropic behaviour of hNSCs toward melanoma cells was also confirmed. The ratio of hNSCs that migrated toward melanoma cells and normal human cells was about 4:1. This migratory effect can be explained by the interaction between chemoattractant factors secreted from melanoma cells and hNSCs. While the melanoma cell line (A375SM) expressed SDF-1 and VEGF; hNSCs expressed the corresponding chemoattractant factors CXCR4 and VEGFR, which suggests that the resulting interactions attracted the hNSCs to the melanoma cells. This tumor-tropic behaviour can mitigate damage to normal tissues and target inaccessible tumor foci.
The therapeutic efficacy of hNSCS engineered with therapeutic genes in the presence of 5-FC was studied on melanoma cells in vitro and in vivo. The melanoma cell viability decreased approximately 50% when co-cultured with HB1.F3.CD or HB1.F3.CD.IFN-β in the presence of 5-FC. However, there was no synergistic effect of HB1.F3.CD.IFN-β on melanoma cells. Melanoma cells were subcutaneously injected on the back of female athymic mice and hNSC treatment was weekly performed for 3 weeks. We injected hNSCs near the tumor site instead of distant sites because we also thought it would be a realistic approach. Melanoma is visible on the skin unlike other cancers, which gives us better application of this therapy by injecting directly at the tumor site; however, if we had injected hNSCs directly at the tumor site, we would not have been able to verify the tumor-tropic migratory behaviour of hNSCs toward melanoma. The therapeutic effect of the HB1.F3.CD and HB1.F3.CD.IFN-β cell line also appeared on xenograft mouse models. The tumor volumes of the HB1.F3.CD and HB1.F3.CD.IFN-β groups were significantly reduced after the 14th day of hNSC treatment, and there was no synergistic effect shown by HB1.F3.CD.IFN-β, which was equivalent to the result of the in vitro cell viability assay. However, the tumor volume of the HB1.F3 group and the group treated only with 5-FC treated constantly increased, and when the tumor volume reached more than 1,500 mm3 necrosis occurred on most of the mice. This necrosis in tumor cells occurs during tumor progression because of the lack of an adequate blood supply for continued growth. The development of necrosis in tumors is more prevalent in aggressive cancers and associated with angiogenesis, hypoxia, and inflammation responses within tumors [26].
The migratory effect of engineered hNSCs was also confirmed in the in vivo study. All three types of hNSCs were pre-labeled with the CM-DiI cell tracker to make them distinct from the tumor tissue before the hNSC treatment was performed on athymic mice. We injected hNSCs at least 1 week prior to the injection of 5-FC which we believe gave enough time for hNSCs to migrate toward tumors and allow conversion of 5-FC to 5-FU occurs. The migration of hNSCs towards tumor foci via the chemoattractant ligand-receptor interactions was shown using fluorescence analysis. The density of CM-DiI pre-labeled hNSCs in the HB1.F3.CD and HB1.F3.CD.IFN-β groups were less than that in the HB1.F3 group. This phenomenon can be explained by the fact that when CD enzyme deaminates the nontoxic drug 5-FC to a toxic drug 5-FU the cell carrying CD gene is eliminated without notable toxicity to the normal cells [24]. The inhibitory effect of engineered hNSCs on melanoma was supported by the in vivo study as well using immunohistochemical analysis. The expression of the cell proliferation gene (PCNA) was diminished for the HB1.F3.CD and HB1.F3.CD.IFN-β groups compared to the vehicle and HB1.F3 groups. The reductions of PCNA expression on tumor growth indicate the therapeutic efficacy of hNSCs treatments in a preclinical experiment.
The active metabolized by-product 5-FU works as a thymidylate synthase inhibitor that promotes apoptosis by binding to a nucleotide-binding sites and destabilizing dNTP in cancer cells [27]. To investigate whether apoptosis-related proteins were activated after the 5-FU treatment, A375SM cells were incubated with different concentrations of 5-FU (1 and 5 μg/mL). The activation of pro-apoptotic protein BAX was increased, and the activation of anti-apoptotic protein Bcl-xL was decreased in the response to 5-FU, which led to cell death. This result implies that 5-FU provokes apoptosis by triggering an imbalance between pro-apoptotic and anti-apoptotic proteins in tumor cells. Moreover, Yi et al. [28] used a breast cell line treated with 5-FU to show that 5-FU exposure both decreases the ratio of Bcl-2/BAX and increases expression of p53 and p38, which led to an increase in apoptosis and repression of cell proliferation through modulation of crucial signaling proteins like p53 and p38. Unlike previous preclinical studies [22,29], we could not verify the synergistic anticancer effect of IFN-β on melanoma. However, the cytokine interferons can not only suppress proliferation of tumor cells but also modify the EMT and mesenchymal to epithelial transition (MET) [30]. To evaluate the function of the recombinant IFN-β on EMT and metastasis, the melanoma cells were cultured with HB1.F3.CD.IFN-β conditioned media. HB1.F3.CD conditioned media was used for comparing the effect of HB1.F3.CD.IFN-β. The expression levels of the epithelial protein E-cadherin, mesenchymal protein Slug, and metastasis protein cathepsin-B were analyzed using a western blot assay. The longer melanoma cells were exposed to IFN-β, the more the process of MET was ameliorated in this study.
Taken together, these results showed that the HB1.F3.CD.IFN-β cell line not only suppressed the proliferation of cancer cells but is also able to promote MET and reduce metastasis, demonstrating that IFN-β from the HB1.F3.CD.IFN-β cell line induced beneficial effects other than prevention of tumor cell proliferation. The functional form of secreted IFN-β will be quantified for the HB1.F3.CD and HB1.F3.CD.IFN-β cell lines using the ELISA method in a further study. Overall, this study elucidated that engineered neural stem cell is an auspicious drug-delivery vehicle for malignant melanoma treatment and could be pragmatic for a cancer-targeted gene therapy.