Introduction
Anaplastic large cell lymphoma (ALCL) is a rare aggressive form of mature T-cell lymphoma, which accounts for approximately 3% of adult non-Hodgkin lymphoma (NHL) and 10%-15% of pediatric NHL [1]. More than half of all ALCL cases harbor an nucleophosmin (NPM)‒anaplastic lymphoma kinase (ALK) fusion gene [2]. The constitutively activated NPM-ALK fusion protein consequently activates a number of signaling pathways such as phosphoinositide 3-kinase (PI3K)/AKT and Ras/ERK [3,4]. Although ALK fusion gene-positive (ALK-positive) ALCL patients have a better outcome than those without the fusion gene, patients with relapsed or refractory disease generally have short survival [1,2]. Crizotinib (also known as PF-02341066) is an ALK tyrosine kinase inhibitor that has an anti-proliferative activity in ALK-positive ALCL cell lines [5]. In patients with lung adenocarcinoma harboring an EML4-ALK fusion gene, crizotinib has shown substantial therapeutic efficacy [6]. It is plausible, therefore, that crizotinib could also be an effective treatment for patients with ALK-positive ALCL.
As one of the crucial downstream signaling molecules of aberrantly activated NPM-ALK, mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase, which consists of mTORC1 and mTORC2. The activity of mTORC1 is regulated by the PI3K/AKT and Ras/ERK pathways [7]. Vega et al. [8] demonstrated that mTOR pathway is frequently activated in ALK-positive ALCL tumors and cell lines. Phosphorylated mTOR was detected in 24 of 29 (82.8%) and phosphorylated p70S6K in 24 of 30 cases (80%), while phosphorylated 4E-BP1 and total eIF4E were both positive in all 31 ALK-positive ALCL tumors examined [8]. Activation of this pathway promotes protein synthesis, capdependent translation of mRNA, angiogenesis, invasion, metastasis, and proliferation of cancer cells. Everolimus (also known as RAD001), an oral rapamycin analog, is an mTOR inhibitor. Jundt et al. [9] demonstrated that everolimus significantly inhibits tumor cell proliferation in ALCL preclinical models, both in vitro and In vivo . Recently, everolimus has also been shown to have clinical activity in patients with diverse NHL subtypes [10]. However, the clinical efficacy of mTOR inhibitor monotherapy has not yet yielded satisfactory results in patients with relapsed or refractory NHL [11], possibly owing to the emergence of resistance mechanisms that activate pro-survival pathways.
Previous studies have shown that crizotinib has limited effect on inhibiting mTOR signaling in ALK-positive neuroblastoma [12]. It is rational to hypothesize that, the strategy of targeting NPM-ALK and mTOR simultaneously might be able to overcome the limitations of each monotherapy. In this study, we investigated whether the combination of crizotinib and everolimus exerts synergistic antitumor activity in ALK-positive ALCL cell lines. The biological mechanism of action of this combination treatment was also investigated.
Materials and Methods
1. Cell lines and culture
Two ALK-positive ALCL cell lines were used in this study: Karpas 299 was obtained from Sigma-Aldrich (St. Louis, MO) and SU-DHL-1 from American Type Culture Collection (Manassas, VA). NCI-H2228, an EML4-ALK–positive lung adenocarcinoma cell line, was obtained from American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (GIBCO, Grand Island, NY) and 1% penicillin/streptomycin (Lonza, Walkersville, MD). All cells were incubated at 37°C in an atmosphere of 5% CO2.
2. Antibodies and reagents
Antibodies specific for ALK, phosphorylated ALK (Tyr1604), phosphorylated mTOR (Ser2448 and Ser2481), phosphorylated AKT (Thr308 and Ser473), phosphorylated ERK (Thr202/Tyr204), phosphorylated p70S6K (Thr389), phosphorylated 4E-BP1 (Thr37/46) and caspase 3 were purchased from Cell Signaling Technology (Beverly, MA). The anti-GAPDH antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). The anti-cleaved poly(ADP-ribose) polymerase (PARP) antibody was purchased from BD Pharmingen (San Jose, CA). The anti-gamma-H2A.X and anti-beta-actin antibodies were purchased from Abcam (Cambridge, UK). The anti-cyclin D1 antibody was purchased from EMD Millipore (Billerica, MA). Everolimus and crizotinib were purchased from Selleckchem (Houston, TX), dissolved in dimethyl sulfoxide and stored at −20°C.
3. Cell viability assays
Cell lines were seeded into 96-well plates and then incubated at 37°C for 72 hours. Cell viability was quantified using the Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan). Following incubation with 10 μL of CCK-8 solution per well for 2 hours, absorbance was measured at 450 nm in a microplate reader (Becton Dickinson Labware, Le Pont de Claix, France). All assays were performed five times. Data were normalized to the percentage of the control.
4. Western blot analysis
Karpas 299 cells were washed in ice-cold phosphate buffered saline (PBS), and then suspended in cell lysis buffer. The mixtures were placed on ice for 1 hour, during which they were vortexed every 10 minutes. After centrifugation for 30 minutes at 13,000 rpm at 4°C to remove cellular debris, the supernatant was collected. Protein quantification was performed using the Micro BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). The samples were boiled at 100°C for 7 minutes, and 10-20 μg of each sample was loaded onto 6%, 10%, 12%, or 15% polyacrylamide gels. After electrophoresis, gels were electro-transferred onto polyvinylidene difluoride membranes (EMD Millipore). After blocking with 5% skim milk and 5% bovine serum albumin (BSA) in Tris-buffered saline with Tween 20 (TBS-T) for 1 hour, the membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with appropriate secondary antibodies for 1 hour the next day. A WEST-ZOL (plus) western blot detection system (Intron, Seongnam, Korea) was used to develop the blot, detected by Medical X-Ray Film Blue from AGFA (Septestraat, Mortsel, Belgium). The data was quantified by Image Studio Lite software ver. 5.2 (LI-COR, Lincoln, NE) and normalized by corresponding GAPDH or β-actin control.
5. Cell-cycle analysis
The cell-cycle distribution was determined by propidium iodide (PI) staining. Karpas 299 cells were treated with everolimus and/or crizotinib. After 72 hours, cells were collected, washed with PBS, and fixed in ice-cold 70% ethanol for at least 2 days but less than 1 week. After staining with 42 μg/mL PI (Sigma-Aldrich), 140 ng/mL RNase A (Invitrogen, Carlsbad, CA), 0.1% BSA, and PBS, the cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Results are expressed as the mean value of five independent experiments.
6. Apoptosis assays
After the cells were exposed to chemotherapeutic agents, the degree of apoptosis was assessed using Annexin V-FITC and PI double-staining according to the protocols of the manufacturer (BD Biosciences). The cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences). Annexin V-positive cells were defined as apoptotic. Results are expressed as the mean value of three independent experiments.
7. In vivo tumor model
For In vivo experiments, 8- to 9-week-old NOD.Cg-Prkdc severe combined immunodeficiency Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory, Bar Harbor, ME) were injected subcutaneously with 1×107 Karpas 299 or SU-DHL-1 cells. When the tumor reached 40 mm3, mice were randomized to four groups of two mice each: control group (saline with dimethyl sulfoxide), everolimus alone (0.25 mg/kg/day), crizotinib alone (10 mg/kg/day), or combination, administered orally once daily for 18 days. Body weight was monitored weekly throughout the experiment until euthanasia. Estimation of volumes by caliper measurement was performed weekly, using the following formula: volume=(width)2×(length)/2. All animal experiments were approved by the institutional animal care and use committee.
8. Immunohistochemistry
Fresh mice xenograft tumors were fixed in formalin and embedded in paraffin. For immunohistochemistry staining, tissue sections of 4 μm in thickness were deparafinized, rehydrated and washed. Each tissue sample was analyzed after hematoxylin and eosin (H&E) staining. Then, apoptosis was analyzed by terminal deoxynucleotidyl transferase–mediated dUTP nick end (TUNEL) assay, using the In Situ Cell Death Detection Kit (Roche, Basel, Switzerland). Monoclonal anti-Ki-67 antibody (clone D2H10) from Cell Signaling Technology (Beverly, MA) was used to detect proliferating cells. All procedures were carried out according to the manufacturer´s recommendation.
9. Statistical analysis
Experimental data were expressed as the mean±standard error. The combination index (CI) was calculated by CalcuSyn software ver. 2.11 (Biosoft, Cambridge, UK) using the median effect methods. A CI of 1 indicates an additive effect of two drugs, less than 1 indicates synergism, and more than 1 indicates antagonism. Statistical differences between the two groups were analyzed using a Student’s t-test for both in vitro and In vivo experiments. All statistical tests were two-sided, with significance defined as p < 0.05. All statistical analyses were performed using Prism 7 (GraphPad Software, La Jolla, CA).
Results
1. Crizotinib combined with everolimus synergistically inhibits proliferation of ALK-positive ALCL cells
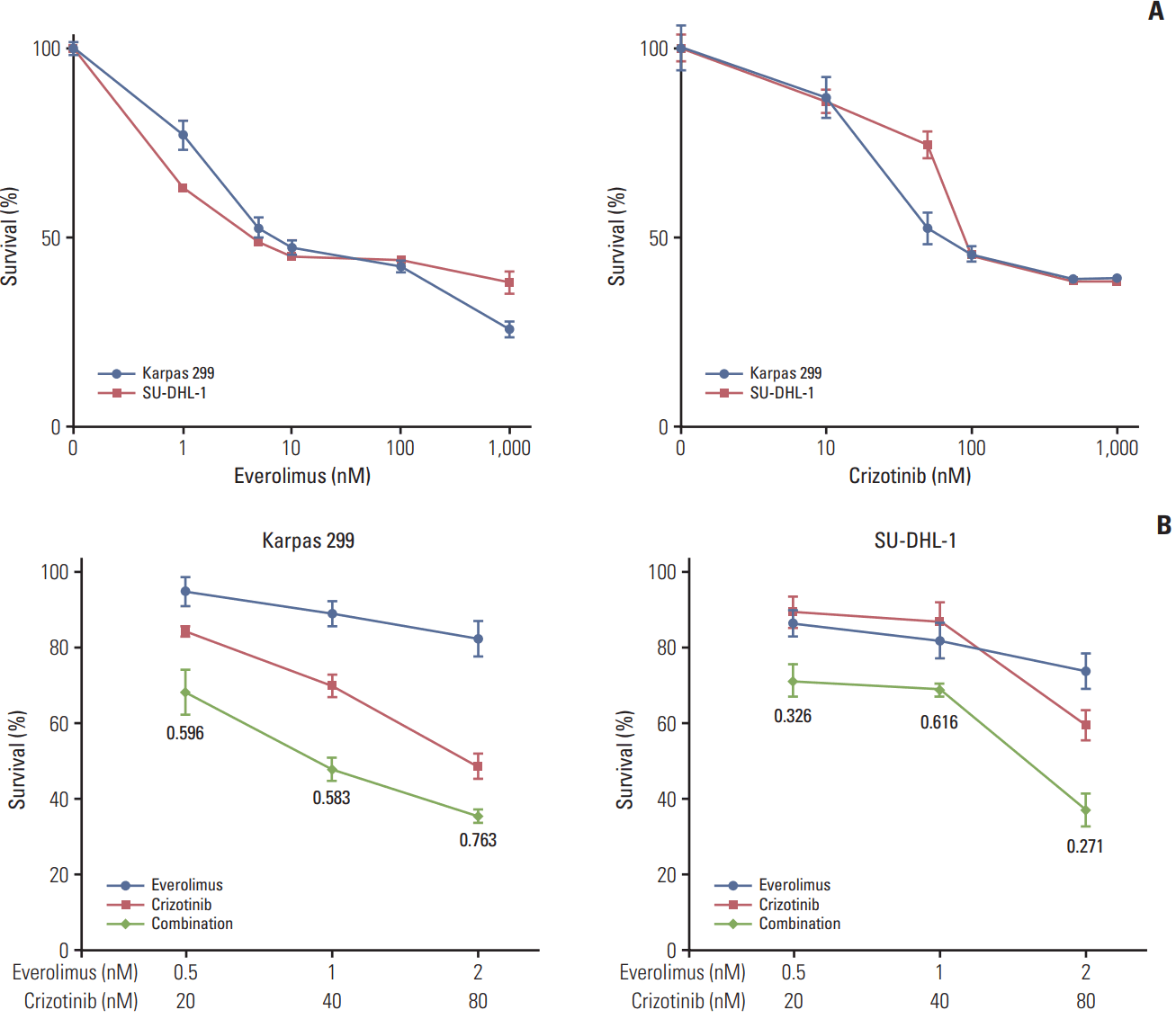
Karpas 299 and SU-DHL-1 cells were treated with 0.001-1 μM everolimus or 0.01-1 μM crizotinib for 72 hours and cell viability was assessed. Both everolimus and crizotinib potently inhibited proliferation in a dose-dependent manner in the two cell lines (Fig. 1A). The half maximal inhibitory concentration (IC50) values for everolimus were 3.8 nM in Karpas 299 and 2.7 nM in SU-DHL-1. IC50 values for crizotinib were 54.1 nM in Karpas 299 and 53.4 nM in SU-DHL-1. Karpas 299 and SU-DHL-1 cells were then treated with everolimus or crizotinib alone, and a combination of the two agents at a fixed ratio of 1:40. The combination of everolimus and crizotinib showed synergistic cytotoxic effects in the two ALCL cell lines compared with everolimus or crizotinib alone (Fig. 1B).
2. Everolimus and crizotinib have different effects on signal transduction pathways
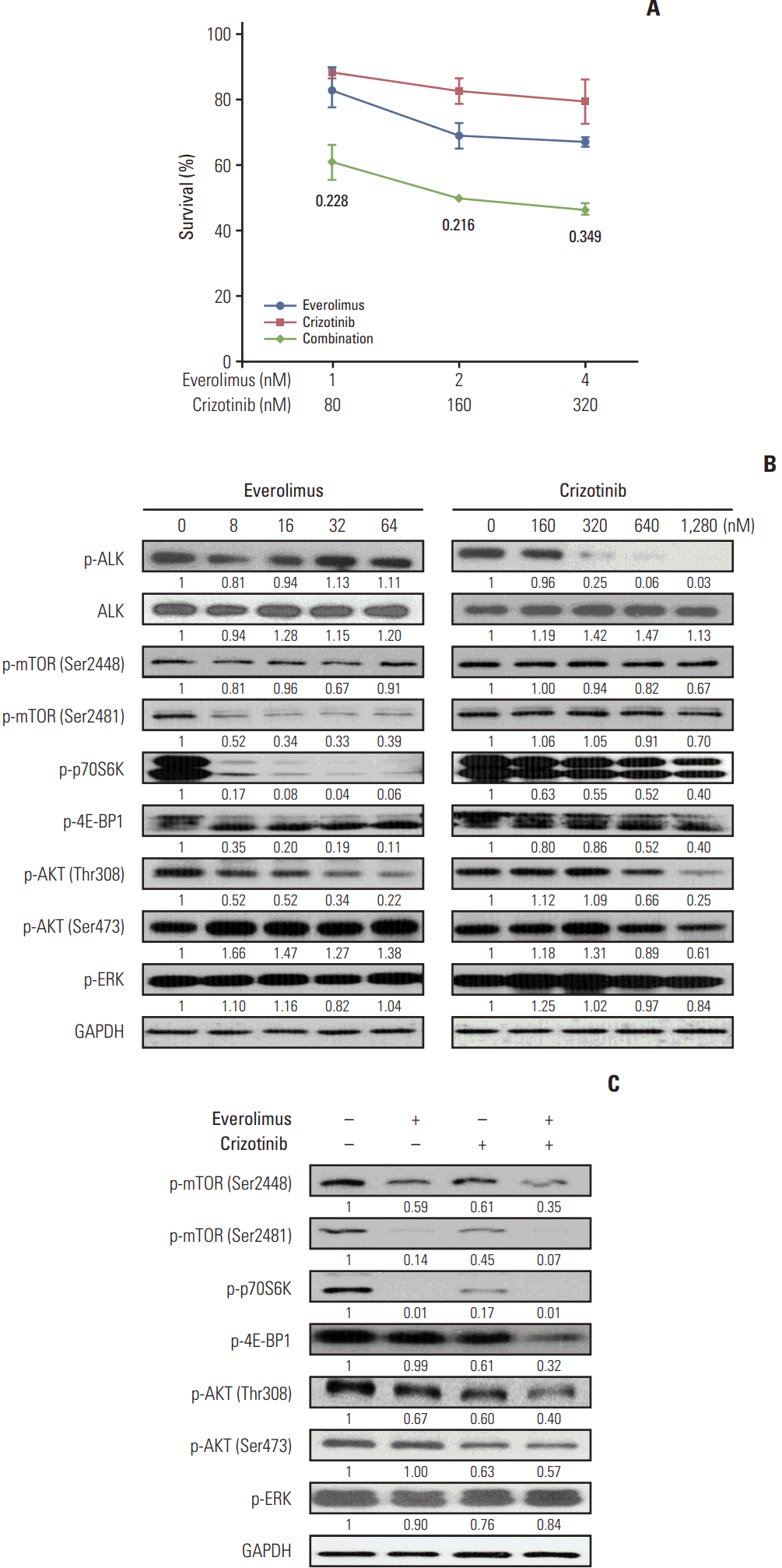
Crizotinib, not everolimus, decreased ALK phosphorylation in Karpas 299 and SU-DHL-1 cell lines, whereas there were no significant effects of crizotinib or everolimus on total ALK expression levels (Fig. 2A). Previous studies have shown that mTORC1 activity can be evaluated by measuring the phosphorylation of Ser2448 on mTOR, while mTORC2 activity may be quantified by assessing the phosphorylation of Ser2481 [13]. Thus, we determined that the activity of mTORC1 and mTORC2 was decreased by everolimus and crizotinib in both Karpas 299 and SU-DHL-1 cells. Phosphorylation of p70S6K was also down-regulated by both everolimus and crizotinib exposure. In Karpas 299 cells, 4E-BP1 phosphorylation tended to increase after everolimus or crizotinib treatment, whereas it tended to decrease in SU-DHL-1 cells. Everolimus resulted in increased phosphorylation of AKT in both cells. In addition, everolimus up-regulated phosphorylated ERK in Karpas 299 cells. After crizotinib treatment, both AKT and ERK phosphorylation was down-regulated in a dose-dependent manner.
3. Crizotinib down-regulates everolimus-induced AKT and ERK phosphorylation and strengthens mTOR inhibition
Karpas 299 and SU-DHL-1 were treated with 2 nM of everolimus and 80 nM crizotinib for 24 hours, separately and in combination (Fig. 2B). In Karpas 299 cells, the combined dose of everolimus and crizotinib further inhibited AKT Thr308 phosphorylation compared with everolimus or crizotinib alone. In SU-DHL-1 cells, the combination treatment further down-regulated phosphorylation of AKT Thr308 and Ser473 and ERK. The combination also decreased phosphorylation of mTOR Ser2448 and Ser2481, and p70S6K Thr389. Decreased mTOR phosphorylation indicated that everolimus combined with crizotinib inhibits both mTORC1 and mTORC2 activities. Phosphorylation of 4E-BP1, another substrate of mTORC1, decreased in SU-DHL-1 cells on exposure to combination treatment, but this decrease was not apparent in Karpas 299 cells.
4. Combined effects of everolimus and crizotinib are reproduced in an ALK-positive lung adenocarcinoma cell line, NCI-H2228
NCI-H2228 cells were treated with everolimus and/or crizotinib at a fixed ratio of 1:80 for 72 hours, and then cell viability was assessed. As expected, crizotinib combined with everolimus exhibited synergistic cytotoxicity (Fig. 3A). Western blot analysis indicated that crizotinib, not everolimus, decreased ALK phosphorylation, whereas there were no significant effects of crizotinib or everolimus on total ALK expression levels (Fig. 3B). Everolimus predominantly down-regulated mTORC2 activity, while crizotinib had little effect. Everolimus potently reduced the phosphorylation of p70S6K and 4E-BP1, while a modest dose-dependent inhibition of p70S6K and 4E-BP1 was observed in crizotinib-treated cells. Expectedly, everolimus tended to increase AKT Ser473 phosphorylation, which was decreased by crizotinib treatment. Both everolimus and crizotinib had little effect on ERK phosphorylation. NCI-H2228 cells were then treated with 16 nM everolimus and/or 1,280 nM crizotinib, alone and in combination, for 24 hours. A combined dose of everolimus and crizotinib effectively decreased phosphorylation of mTOR, p70S6K, 4E-BP1, and AKT (Fig. 3C). ERK phosphorylation, however, was barely altered after the combination treatment.
5. When combined, crizotinib and everolimus induce cellcycle arrest and apoptosis
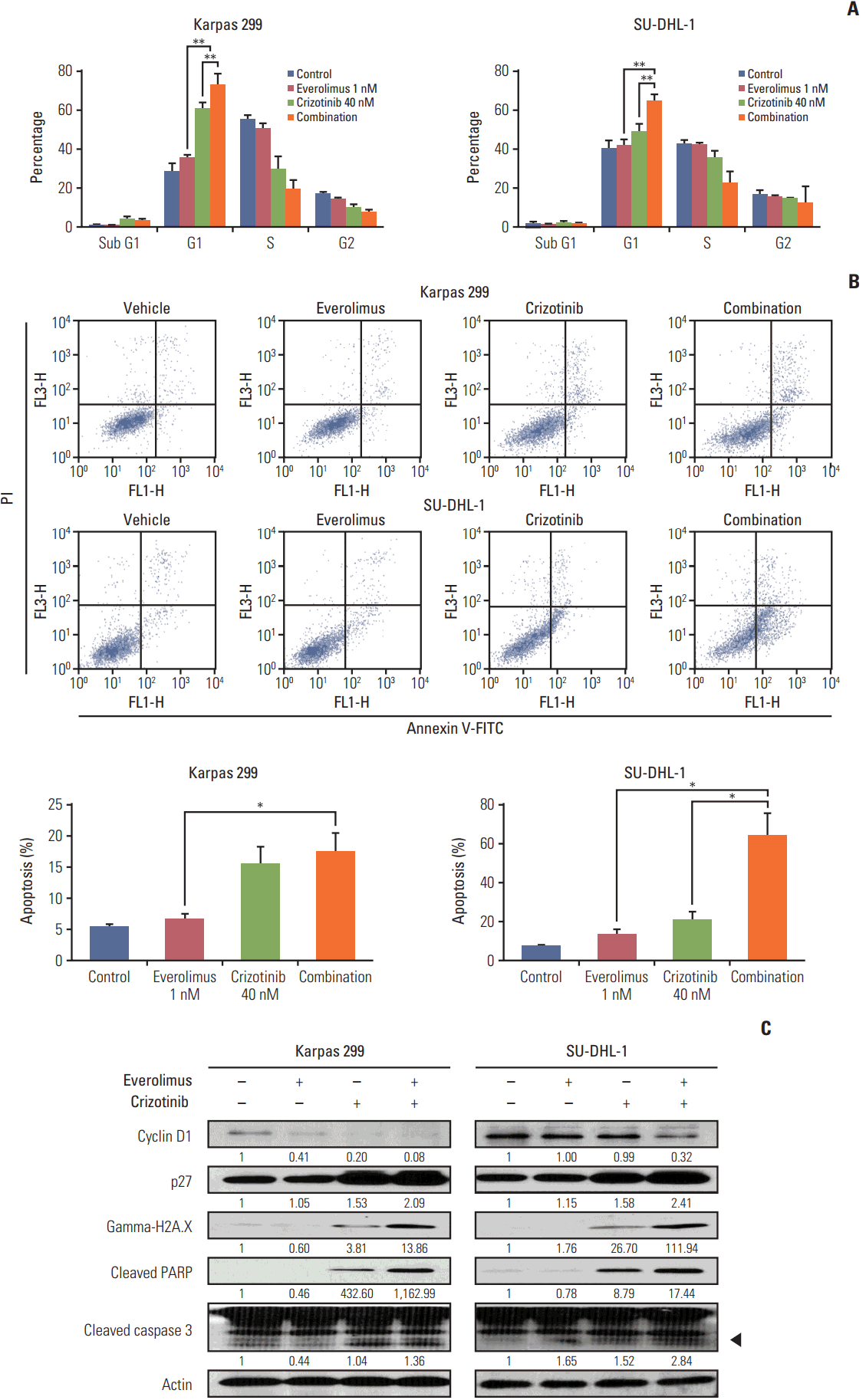
Karpas 299 and SU-DHL-1 cells were treated with 1 nM everolimus and 40 nM crizotinib, alone and in combination, for 72 hours, after which we performed PI staining and flow cytometry (Fig. 4A). Crizotinib as a single agent significantly increased cell-cycle arrest in G0/G1 phase (28.25±1.19% vs. 60.48±1.93% in Karpas 299 cells [p < 0.001] and 40.36±1.39% vs. 48.88±1.52% in SU-DHL-1 cells [p=0.003]). Everolimus alone significantly increased G0/G1 cell-cycle arrest in Karpas 299 cells (28.25±1.19% vs. 35.26±0.96%, p=0.002), and SU-DHL-1 cells, although the increase in SU-DHL-1 was not significant (40.36±1.39% vs. 41.91±0.90%, p=0.378). Importantly, the fraction of cells blocked in G0/G1 phase was significantly increased by the combination treatment compared with crizotinib alone (60.48±1.93% vs. 72.83±2.08% in Karpas 299 cells [p=0.002] and 48.88±1.52% vs. 64.83±1.57% in SU-DHL-1 cells [p < 0.001]).
Karpas 299 and SU-DHL cells were treated with 1 nM everolimus and 40 nM crizotinib, alone or in combination, for 72 hours, and then stained with Annexin V‒FITC and PI for apoptosis analysis by flow cytometry (Fig. 4B). Everolimus tended to increase apoptosis compared with the control, although this was not significant (5.36±0.50% vs. 6.60±0.98% in Karpas 299 cells [p=0.322] and 7.45±0.64% vs. 13.16±2.81% in SU-DHL-1 cells [p=0.118]). Crizotinib as a single agent increased apoptosis (5.36±0.50% vs. 15.39±2.77% in Karpas 299 cells [p=0.023] and 7.45±0.64% vs. 20.29±4.59% in SU-DHL-1 cells [p=0.0503]). Compared with everolimus alone, the combination treatment further increased apoptosis in both Karpas 299 (6.60±0.98% vs. 17.38±2.99%, p=0.027) and SU-DHL-1 cells (13.16±2.81% vs. 64.04±11.64%; p=0.013), to a significant degree. Similarly, when compared with crizotinib alone, the combination treatment significantly increased apoptosis in SU-DHL-1 (20.29±4.59% vs. 64.04±11.64%, p=0.025), and tended towards increasing apoptosis in Karpas 299 cells (15.39±2.77% vs. 17.38±2.99%, p=0.652).
Western blot analysis revealed that the combination treatment down-regulated cyclin D1 and up-regulated p27. In addition, the combination treatment promoted cleavage of PARP and caspase 3, and increased gamma-H2A.X expression (Fig. 4C).
6. Combination of everolimus and crizotinib shows an enhanced antitumor effect compared to single treatments in the Karpas 299 xenograft model
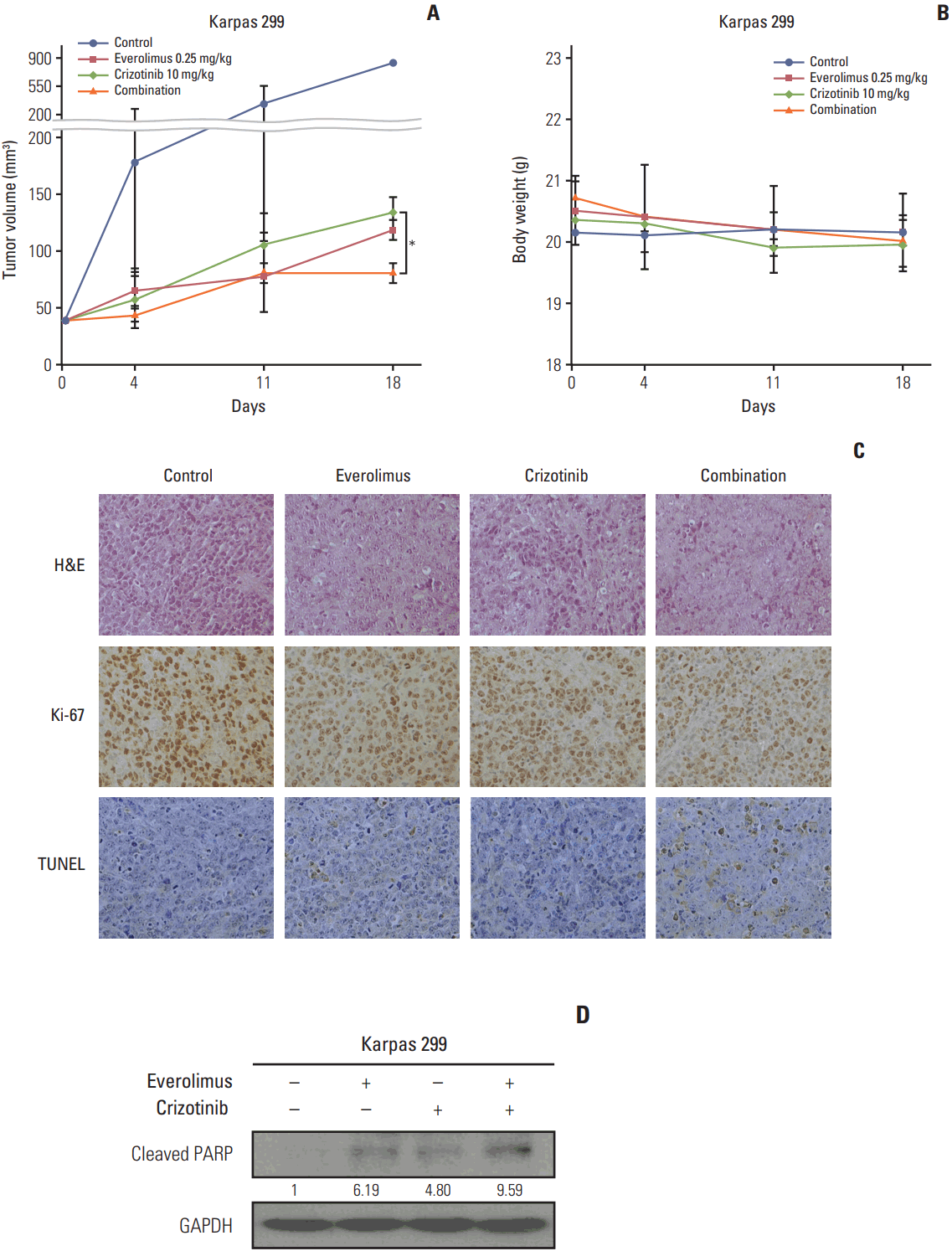
To find out if our in vitro findings could be translated into an In vivo setting, we evaluated the antitumor effects using a mouse xenograft model of Karpas 299 cell line. Everolimus or crizotinib as a single agent exerted a significant antitumor effect (p < 0.001 for both everolimus and crizotinib) (Fig. 5A). The combination treatment of everolimus and crizotinib further decreased tumor volume compared with crizotinib alone (81.25±8.84 mm3 vs. 135.00±12.73 mm3, p=0.039) and everolimus alone (81.25±8.84 mm3 vs. 118.75±8.84 mm3, p=0.051). In addition, there was no significant change in body weight in each treatment group and no significant difference in body weight among treatment groups (Fig. 5B). Similar results were also reproduced in an SU-DHL-1 xenograft model (S1 Fig.).
To evaluate changes in tumors under the effect of everolimus and/or crizotinib, we first stained tumor tissues with H&E. Everolimus or crizotinib alone induced apoptotic cells in tumors evidenced by pyknotic nucleus, in comparison with vehicle-treated control. The tumors with combination treatment showed not only pyknotic cells, but also large areas of dead cells (Fig. 5C). Everolimus combined with crizotinib also decreased Ki-67 expression compared to monotherapy, indicating anti-proliferative activity of the combination treatment. The staining of TUNEL indicates the existence of DNA fragmentation; therefore, the tumor section of combination treatment showed enhanced apoptosis compared to control or single treatment. Western blot analysis also revealed an increased cleaved-PARP after combination treatment (Fig. 5D).
Discussion
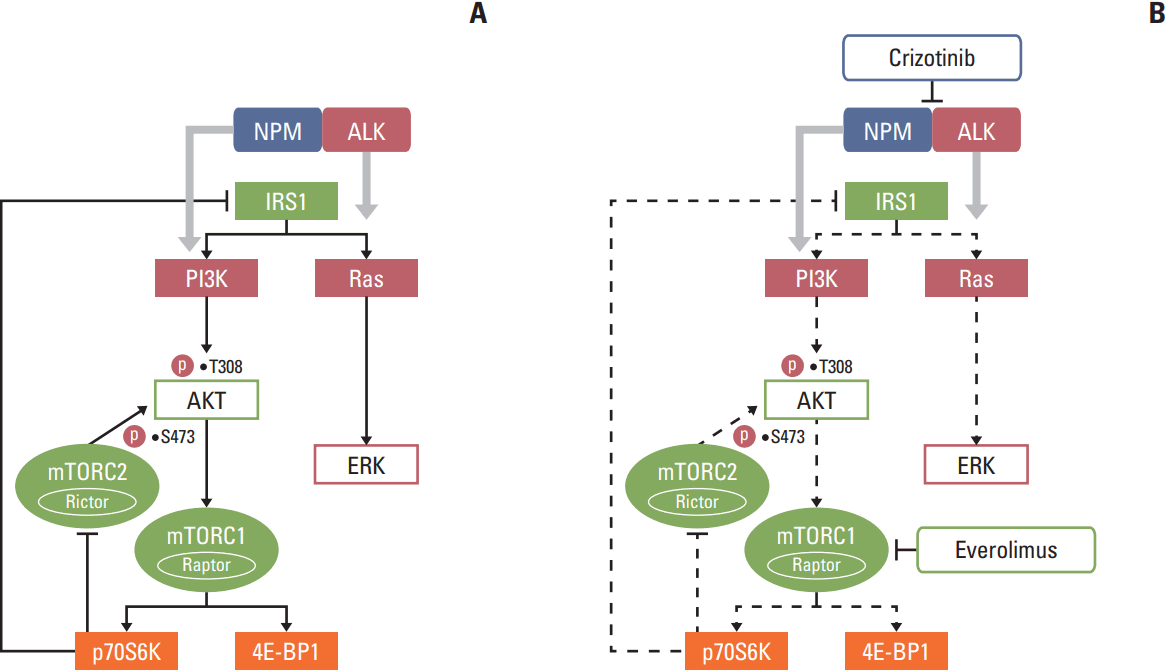
In this study, the combination of everolimus, an mTOR inhibitor, and an ALK inhibitor crizotinib showed a synergistic antitumor activity in ALK-positive preclinical models. We show that everolimus increased AKT and ERK phosphorylation in ALK-positive ALCL cells. Crizotinib in combination with everolimus demonstrated synergistic cytotoxicity by abrogating aberrant AKT and ERK phosphorylation induced by everolimus (Fig. 6). In addition, this combination treatment significantly increased G0/G1 cell-cycle arrest, DNA damage, and apoptosis. Moreover, this anti-proliferative activity could be translated into an ALCL In vivo xenograft model.
The mTOR signaling pathway is frequently deregulated in many human cancers including ALCL [8,14]; thus, it is anticipated that inhibiting mTOR would be a promising strategy for the treatment of a variety of cancers. Currently, there are several mTOR inhibitors under clinical development as anti-cancer therapies. However, previous reports showed that mTORC1 inhibition using rapalogs leads to up-regulation of AKT and ERK by several different mechanisms [15-18]. One of the major substrates of mTORC1, p70S6K, regulates the pathway via a feedback mechanism in which PI3K is inhibited by IRS1 down-regulation. Thus, mTORC1 inhibition using rapalogs, which down-regulates p70S6K, leads to PI3K activation [15]. The p70S6K also inhibits mTORC2 through phosphorylation of Rictor at Thr1135 [16]. Therefore, p70S6K down-regulation by rapalogs allows mTORC2 activity, leading to AKT phosphorylation on Ser473. In addition, mTOR inhibition increases platelet-derived growth factor receptor expression, leading to further increase in AKT phosphorylation on Ser473 [17]. ERK activation is also triggered by mTORC1 inhibition through the p70S6K-IRS-Ras pathway in cancer patients as well as in vitro experiments and a preclinical model of prostate cancer [18]. In our study, crizotinib attenuated AKT and ERK activation triggered by everolimus.
In ALK-positive ALCL cells, the aberrant NPM-ALK fusion gene activates Ras/ERK, STATs, and PI3K/AKT signaling pathways. Crizotinib effectively inhibits phosphorylation of ALK, AKT, ERK, PLC-γ, and STAT3, both in vitro and In vivo [19]. A previous report showed that two ALK-positive ALCL patients could achieve rapid complete remission after crizotinib treatment, even though they had previously relapsed after CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy [20]. Another clinical study of crizotinib monotherapy in patients with relapsed or refractory ALK-positive NHL reported an overall response rate of 91% (10 of 11) with 2-year overall and progression-free survival rates of 72.7% and 63.7%, respectively [21]. However, a substantial portion of patients eventually experienced relapse or progression despite crizotinib treatment, possibly due to acquired resistance to crizotinib developed during treatment. In previous preclinical crizotinib study in ALK-positive ALCL, Christensen et al. [19] demonstrated that crizotinib was ineffective at downregulating phosphorylated AKT, a main up-stream molecule of mTOR, even at doses that inhibited phosphorylated ALK completely. Berry et al. [12] demonstrated that while crizotinib abolished ALK phosphorylation, it had little effect on AKT, S6K and 4EBP1 phosphorylation in ALKF1174L/MYCN neuroblastoma model, which leads to a conclusion that the resistance to crizotinib might correlate to incomplete inhibition of key signaling pathways including mTOR. Therefore, we hypothesized that, rather than a monotherapy, crizotinib might be used more effectively as a part of a combination therapy with other chemotherapeutic agents.
As for the mechanism of action, crizotinib binds to the ATP binding site of ALK enzyme and subsequently prevents auto-phosphorylation. In line with our western blot results, Megiorni et al. [22] demonstrated that crizotinib treatment inhibited the phosphorylation of ALK, AKT, and ERK in rhabdomyosarcoma cells. However, there was no change in total protein expression levels of these molecules in relation to crizotinib treatment [22]. As for everolimus, it acts by binding to the immunophilin FK binding protein-12, and thus inhibits the activation of mTOR. In agreement with our experimental data, Xu et al. [23] demonstrated that everolimus treatment in diffuse large B-cell lymphoma cell lines substantially decreased phosphorylation of AKT (Thr308), mTOR (Ser2448), p70S6K, and 4E-BP-1 (Thr37/46), without corresponding changes in total protein expression levels of these molecules. These data suggest that neither of the two targeted agents acts at transcriptional level. Therefore, these two targeted agents exert inhibitory effects on signaling pathways by decreasing phosphorylation of target molecules.
Our results showed that, on one hand, the synergistic antiproliferative effect of this novel combination suggested that ALK inhibition could help overcome everolimus resistance in ALK-positive ALCL cells by inhibiting down-stream PI3K/AKT and Ras/ERK signaling pathways. In addition, the effect of everolimus on mTORC1 and p70S6K down-regulation was strengthened by the addition of crizotinib. On the other hand, crizotinib had less effect on mTOR phosphorylation, especially on Ser2448, which is a down-stream target of AKT. This limitation of crizotinib could be overcome by addition of everolimus. Everolimus added to crizotinib further decreased mTOR phosphorylation uniformly in the three ALK-positive cell lines we tested. Recently, a synergistic effect between crizotinib and temsirolimus, another mTOR inhibitor, in ALK-positive ALCL cell lines has been reported by Redaelli et al. [24]. In their study, the combination treatment inhibited tumor growth both in vitro and In vivo, along with a sharp cell-cycle block in G0/G1 phase, which is in agreement with our study. Moreover, our study highlighted the detailed mechanisms that the synergistic antitumor effects of ALK and mTOR inhibitors were mediated by enhanced apoptosis due to the effective inhibition of both up-stream and down-stream of mTOR signaling. These results suggest that the limitations of anti-ALK or anti-mTOR monotherapy could be overcome by the combination treatment. Nevertheless, the effect of everolimus or crizotinib on 4E-BP1 phosphorylation was not consistent in the cell lines tested. A previous study showed that rapamycin could induce 4E-BP1 phosphorylation within 6 hours despite initial inhibition within 3 hours [25]. Crizotinib can also induce 4E-BP1 phosphorylation in ALK-positive lung cancer cells although the precise mechanism remains unknown [26]. The combination of crizotinib and everolimus did not change 4E-BP1 phosphorylation in Karpas 299 cells, whereas it markedly reduced 4E-BP1 phosphorylation in SU-DHL-1 and NCI-H2228 cells (Figs. 2B and 3C). In previous studies performed using lung cancer cells, rapamycin-induced 4E-BP1 phosphorylation could be abrogated by BKM120 (a PI3K inhibitor) or BEZ235 (a PI3K/mTOR dual kinase inhibitor) [27,28]. Therefore, it is hypothesized that PI3K inhibition might be applicable to overcome the limitation of crizotinib and everolimus combination therapy.
Previous studies have shown that crizotinib induces G0/G1 arrest in ALK-positive ALCL cells [19], which is in keeping with our results. Notably, the addition of everolimus to crizotinib significantly increased the G0/G1 population. In accordance with this finding, cyclin D1 was markedly down-regulated by the combination treatment. In the present study and in previous reports, both rapalog and crizotinib induced apoptosis in ALK-positive ALCL cells [19,29]. In our study, the combination of crizotinib and everolimus significantly increased the Annexin V‒stained population in both Karpas 299 and SU-DHL-1 cells (Fig. 4B). In addition, the combination increased gamma-H2A.X expression and cleavage of PARP and caspase 3 (Fig. 4C), consistent with increased DNA damage and apoptosis. An enhanced apoptosis was also observed in a Karpas 299 xenograft model after the combination treatment, as evidenced by stronger TUNEL staining in immunohistochemistry and increased PARP cleavage in western blot. Taken together, our results suggest that the combination treatment promotes G0/G1 cell-cycle arrest, DNA damage, and apoptosis.
Several ALK-fusion genes have been identified in a variety of cancers, such as lung adenocarcinoma, inflammatory myofibroblastic tumor, and renal cell carcinoma [30]. Therefore, we speculate that our finding could be applied to other cancers harboring ALK-fusion genes. We tested this hypothesis in an ALK-positive lung adenocarcinoma cell line. The combination of crizotinib and everolimus showed a synergistic cytotoxic effect in NCI-H2228 cells (Fig. 3). In addition, the combination treatment effectively abrogated the PI3K/AKT and mTOR signaling pathways, with little effect on the Ras/ERK signaling pathway. This result suggests that the combination might be an effective treatment strategy in other cancers harboring ALK-fusion genes.
Our in vitro results suggest that the combination of everolimus and crizotinib demonstrated synergistic anti-proliferative activity through inhibition of down-stream signaling pathways of ALK. However, the potential of clinical translation of this novel combination encourages an In vivo validation. In our Karpas 299 murine model, an enhanced reduction in tumor volume was observed in the combination treatment group, as compared with the single-agent treatment groups, without significant change in body weight of mice.
The combination of crizotinib and everolimus synergistically inhibited proliferation of ALK-positive ALCL cells in both in vitro and In vivo experiments. Crizotinib inhibited aberrant AKT and ERK phosphorylation induced by everolimus, and more effectively suppressed the activity of mTOR than everolimus monotherapy, resulting in increased G0/G1 cell-cycle arrest, DNA damage, and apoptosis. Further clinical trials using crizotinib and everolimus combination are therefore warranted in ALK-positive ALCL. We suggest that this novel combination could be used to improve therapeutic outcome in patients with ALK-positive ALCL.