Introduction
The treatment of cancer stem cells (CSCs) is still a challenge in the management of cancer in humans. Characteristics, such as chemo- and radio-resistance [1], lead to persistence in patients and is a known cause for drug refractory relapse. CD133 has been identified as a frequently expressed marker for CSCs in different tumor types [2,3]. CD133, a transmembrane glycoprotein, is expressed on the cell surface of normal stem cells as well as on CSCs, but CSCs may be characterized by a higher CD133 copy number, providing a basis for effective targeting [4]. CD133 has an important clinical impact in cancer through its association with the Wnt/β-catenin pathway and has demonstrated prognostic relevance in cancer patients [5].
Natural killer (NK) cells are large granular lymphocytes that are known for their potential to recognize malignanttransformed and virus-infected cells via a sophisticated repertoire of activating and inhibitory surface receptors that mediate the killing of the target by the insertion of cytotoxic perforin and granzyme toxins. These characteristics make them important effectors of immune surveillance and cancer defense. The lymphoid repertoire of the human body comprises several NK cell subpopulations that differ in their level of receptor expression. The main population capable of eliminating tumor cells is composed of CD56dimCD16bright cells. The mechanisms for NK-mediated tumor elimination include natural cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC), and the secretion of interferon (IFN)-γ, which are essential for mediating the anti-tumor activity.
NK cells are activated by interleukin (IL)-15 [6] by binding to the IL-15 receptor complex (3 subunits, 2 of them shared with the IL-2 receptor complex). IL-15 mediates the lymphokine-activated killer activity and increases ADCC [6-8]. IL-15 and IL-2 have the ability to mediate the ex vivo expansion of NK cells; up to 20-fold expansion after 2 weeks [9]. This cytokine may have certain therapeutic advantages over IL-2. Studies have shown the benefits of the expansion of allogeneic peripheral blood mononuclear cells (PBMCs) for cancer therapy, but the approach is limited by T cell contamination and the induction of graft-versus-host disease. The potential of IL-15 to induce the expansion and prolonged survival of NK cells in vivo indicates the potential to improve NK cell performance and implicates its use as an effective immunotherapeutic agent [10], particularly if delivered selectively to the NK cell population and the exact site of ADCC in vivo.
Several immunotherapeutic agents were reported to target CD16 [11,12]. All of these show the capability of targeting CD16 target cells selectively. On the other hand, none of these have incorporated IL-15 as a self-sustaining agent capable of stimulating NK expansion.
This study presents a self-sustaining trispecific NK cell engager (1615133 TriKE) that specifically kills CD133+ target cells through the mechanism of ADCC. The construct is composed of a scFv isolated from a hybridoma generated by immunizing against the extracellular domain of CD133. 1615133 TriKE recognizes a framework determinant that crosses species platforms [13]. The construct also contains an anti-CD16 scFv generated in a human phage display library [11] that binds FcGRIII on NK cells to form an immune synapse between the NK cells and CD133 expressing tumor targets, which results in NK cell activation and directed tumor target killing. A human IL-15 encoding cross linker that enhances anti-cancer reactivity significantly was included to provide a priming and expansion signal, leading to an improvement in the NK cell therapeutic potential. This new platform technology has been shown to work with other scFvs targeting a range of cancer antigens and it is hoped that this new iteration will facilitate the targeting of the CSC pool [14-16].
Materials and Methods
1. Construction of 1615133 TriKE
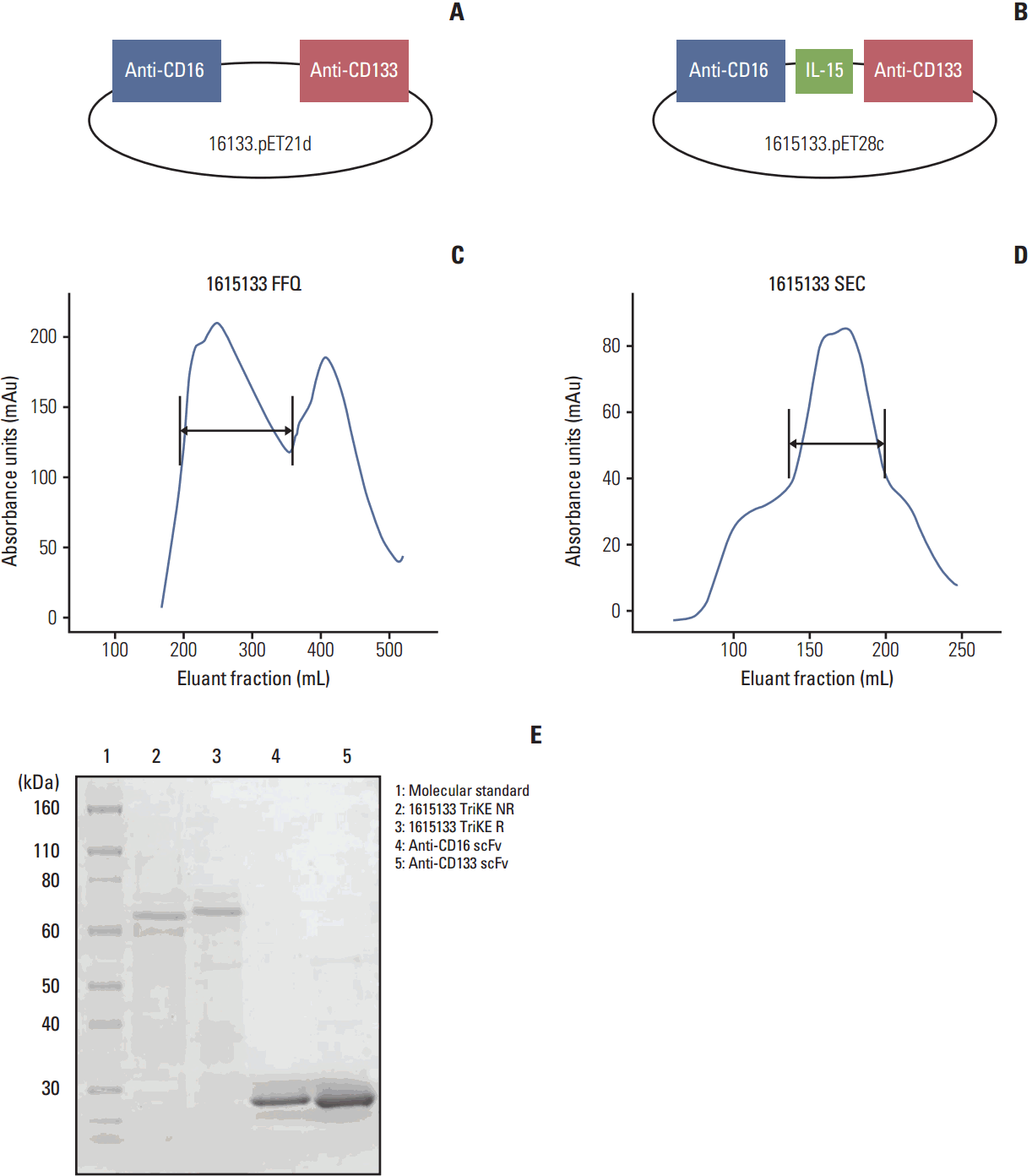
The hybrid gene encoding 1615133 was synthesized using DNA shuffling and DNA ligation techniques. The fully assembled gene (from 5′ end to 3′ end) encoded a NcoI restriction site; an ATG start codon; anti-human CD16 scFv [17]; a 20 amino acid (aa) segment, PSGQAGAAASESLFVSNHAY; N72D-mutated human IL-15 [18]; the seven amino acid linker, EASGGPE; anti-CD133 scFv [13]; and a NotI restriction site. The resulting 1,884 base pair NcoI/NotI fragment gene was spliced into the pET28c expression vector under the control of an isopropyl-β-D-thiogalactopyranoside (IPTG) inducible T7 promoter. DNA sequencing analysis (Biomedical Genomics Center, University of Minnesota, St. Paul, MN) was used to verify that the gene sequence was correct and had been cloned in frame.
2. Isolation of inclusion body
For the expression of proteins, the Escherichia coli strain BL21 (DE3) (Novagen, Madison, WI) was used after plasmid transfection. The bacteria were cultured overnight and grown in 800-mL Luria broth containing 50 mg/mL carbenicillin. When the media reached an absorbance of 0.65 at 600 nm gene expression was induced via the addition of IPTG (FischerBiotech, Fair Lawn, NJ). After 2 hours, the bacteria were harvested. After a homogenization step in a buffer solution (50 mM Tris, 50 mM NaCl, and 5 mM EDTA pH 8.0), the pellet was sonicated and centrifuged. To extract the pellet, 0.3% sodium deoxycholate, 5% Triton X-100, 10% glycerin, 50 mmol/L Tris, 50 mmol/L NaCl, and 5 mmol/L EDTA (pH 8.0), was used and the extract was washed.
3. Refolding and purification
Refolding and purification processes were described recently [19,20]. To refold the proteins, inclusion bodies were dissolved at a 20:1 ratio in a solubilization buffer (7 M guanidine hydrochloride, 50 mM Tris, 50 mM NaCl, 5 mM ethylenediaminetetraacetic acid, and 50 mM dithiothreitol, pH 8.0) and incubated for 1 hour at 37°C. After incubation, the pellets were removed by centrifugation and the supernatant was diluted (20-fold) with a refolding buffer (50 mM Tris-HCl, 50 mM NaCl, 0.8 mM L-arginine, 20% glycerin, 5 mM EDTA, and 1 mM GSSG at pH 8.0) and incubated further for 2 days at 4°C. To remove the buffer, 10-fold dialysis against 20 mM Tris-HCl at pH 9.0 in 20 mM Tris-HCl at pH 9.0 over four column volumes was used. To evaluate the purity, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using Simply Blue life Stain (Invitrogen, Carlsbad, CA). The size of 1615133 was approximately 66,680 Da.
4. Tissue culture
The following cell lines were obtained from the American Type Culture Collection: colorectal carcinoma cell line, Caco-2; and Burkitt lymphoma cell line, Raji. The Caco-2 cell line was grown in monolayers [21] using RPMI supplemented with 20% fetal bovine serum (FBS) and 2 mmol/L L-glutamine. Raji was grown in suspension using Eagle’s minimum essential medium supplemented with 20% FBS and glutamine. Both cell lines were incubated at a humidified atmosphere containing 5% CO2 at a constant 37°C. When the adherent cells were more than 90% confluent, they were passaged using trypsin-EDTA for detachment. For the cell counts a standard hemocytometer was used. Only those cells with a viability > 95% were used for the experiments, as determined by trypan blue exclusion.
5. Isolation of NK cells and purification
To isolate the PBMCs, a histopaque gradient (Sigma-Aldrich, St. Louis, MO) and SepMate tubes (Stemcell Technologies, Vancouver, Canada) were used. Adult blood was derived from healthy volunteers (Memorial Blood Center, Minneapolis, MN). To enrich the NK cells, magnetic beads (Stemcell Technologies) were used according to the manufacturer´s protocol by performing a negative selection. The purity was determined by flow cytometry. The samples were obtained after informed consent and in accordance with the University of Minnesota human subjects Institutional Review Board and the Declaration of Helsinki.
6. Proliferation assay
The PBMCs or enriched NK cells from healthy donors were labeled with a proliferation dye (CellTrace Violet Cell Proliferation Dye, Invitrogen) according to the manufacturer’s protocol. After staining, the effectors were cultured with 50 nM of the respective drugs (anti-CD16 scFv, anti-CD133 scFv, 16133 bispecific NK cell engager (BiKE), and 1615133 TriKE; a targeted toxin consisting of an anti-CD22 and anti-CD19 scFv linked to a diphtheria toxin [DT2219]; or National Cancer Institute [NCI] derived IL-15 alone). After incubation for 7 days in a humidified atmosphere containing 5% CO2 at 37°C, the cells were harvested, stained for their viability with Live/Dead reagent (Invitrogen), and surface stained for anti-CD56 PE/Cy7 (BioLegend, San Diego, CA) and anti-CD3 PE-CF594 (BD Biosciences, Franklin Lakes, NJ) to gate on the viable CD3–CD56+ population. Data analysis was performed using FlowJo software ver. 7.6.5. (Flowjo Enterprise LCC, Ashland, OR) and proliferation was characterized using the expansion index readout. The formula for the expansion index is described elsewhere [22].
7. CD107a degranulation assay
Flow cytometry assays to quantify the lytic degranulation via CD107a surface expression and intracellular IFN-γ presence were reported previously [23]. The PBMCs were incubated over-night (37°C, 5% CO2) in RPMI 1640 media supplemented with 10% fetal calf serum (RPMI-10). The positive control was supplemented further with 10 ng/mL recombinant IL-12 (Peprotech, Rocky Hill, NJ) and 100 ng/mL IL-18 (R&D Systems, Minneapolis, MN). PBMCs were resuspended with noted targets or media after several washing steps with RPMI-10, the cells were then exposed to 50 nM of 1615133 TriKE or the other drugs (16133 BiKE, NCI derived IL-15, anti-CD16 scFv, anti-CD133 scFv) and incubated for 10 minutes at 37ºC with 5% CO2. Fluorescein isothiocyate (FITC)–conjugated anti-human CD107a monoclonal antibody (mAb) (lysosomal-associated membrane protein 1) (BD Biosciences, New Jersey, CA) was then added and incubated for 1 hour. After incubation, GolgiStop (1:1,500, BD Biosciences, San Jose, CA) and GolgiPlug (1:1,000, BD Biosciences, San Jose, CA) were added for 3 hours (37°C, 5% CO2). After the washing steps with 1× phosphate buffered saline, the cells were stained with PE/Cy 7-conjugated anti-CD56 mAb, APC/Cy 7-conjugated anti-CD16 mAb, and PE-CF594–conjugated anti-CD3 mAb (BioLegend). The cells were then incubated for 15 minutes at 4°C, washed and fixed with 2% para-formaldehyde. After the fixing step, the cells were exposed to permeabilization buffer (BD Biosciences, San Jose, CA) and incubated with Pacific Blue–conjugated antihuman IFN-γ (BioLegend) for 20 minutes. The cells were finally washed and evaluated by fluorescence-activated cell sorting analysis using a LSRII flow cytometer (BD Biosciences, San Jose, CA) gating on CD56+CD3– cells.
8. 51Chromium release cytotoxicity assay
The Caco-2 cells were labeled with 1 μCi of 51Cr per 1×105 target cells at 37°C, 5% CO2 for 1 hour. After washing to remove the excess 51Cr, the Caco-2 cells were added to a 96-well round-bottom plate (5×103 cells). The resting PBMCs were treated with 1, 5, and 10 nM 1615133 TriKE; 10 nM 16133 BiKE; or 10 nM anti-CD133 or anti-CD16 scFv at an effector:target (E:T) ratio ranging from 0.08:1 to 20:1. The amount of 51Cr released corresponds to the target cell death and was measured using a gamma scintillation counter. The percentage target cell lysis was calculated using the following formula: [(experimental lysis–spontaneous lysis)/(maximal lysis–spontaneous lysis)]×100. To determine the maximal lysis, the 51Cr-labeled target cells were treated with 3% Triton X for 4 hours.
9. Binding/blocking assay
To evaluate the drug binding, 4×105 Caco-2 cancer cells were washed and incubated in 4°C with 1, 5, 10, 50, 100, 200, and 500 nM of FITC-labeled 1615133 TriKE for 30 minutes. For blocking, 200 nM of an anti-CD133 scFv was added to the same concentrations of the FITC labeled 1615133 and incubated for 30 minutes at 4°C. After washing, the staining intensity was evaluated using an LSRII flow cytometer (BD Biosciences, San Jose, CA).
10. Statistical analyses
The data are presented as the mean±standard deviation. For statistical evaluation a Student’s t test was used to compare two groups, whereas one-way analysis of the variance (ANOVA) was used for multiple comparisons. Data analysis and presentation were performed using Graphpad Prism 5 (GraphPad Software, Inc., La Jolla, CA).
Results
1. 1615133 production and purification
To produce a self-sustaining hybrid NK cell engager, the 16133 BiKE platform (Fig. 1A) was altered with a modified IL-15 crosslinker incorporated between the two scFv constructs forming 1615133 TriKE (Fig. 1B). The new construct contained DNA fragments from the VH and VL regions of an anti-CD16 scFv, an N72D-modified IL-15 flanked with a 20 aa segment and the 7 aa linker EASGGPE, and the VH and VL regions of an anti-CD133 scFv. Fig. 1C presents the absorbance tracing from the FFQ ion exchange column as the first phase in drug purification. The first major peak eluted from the column represents the drug. Size exclusion column purification (Fig. 1D) and SDS-PAGE gel (Fig. 1E) with Coomasie Blue staining showed high purity (over 90%) and a protein size of 66,680 Da.
2. Activity of the IL-15 moiety
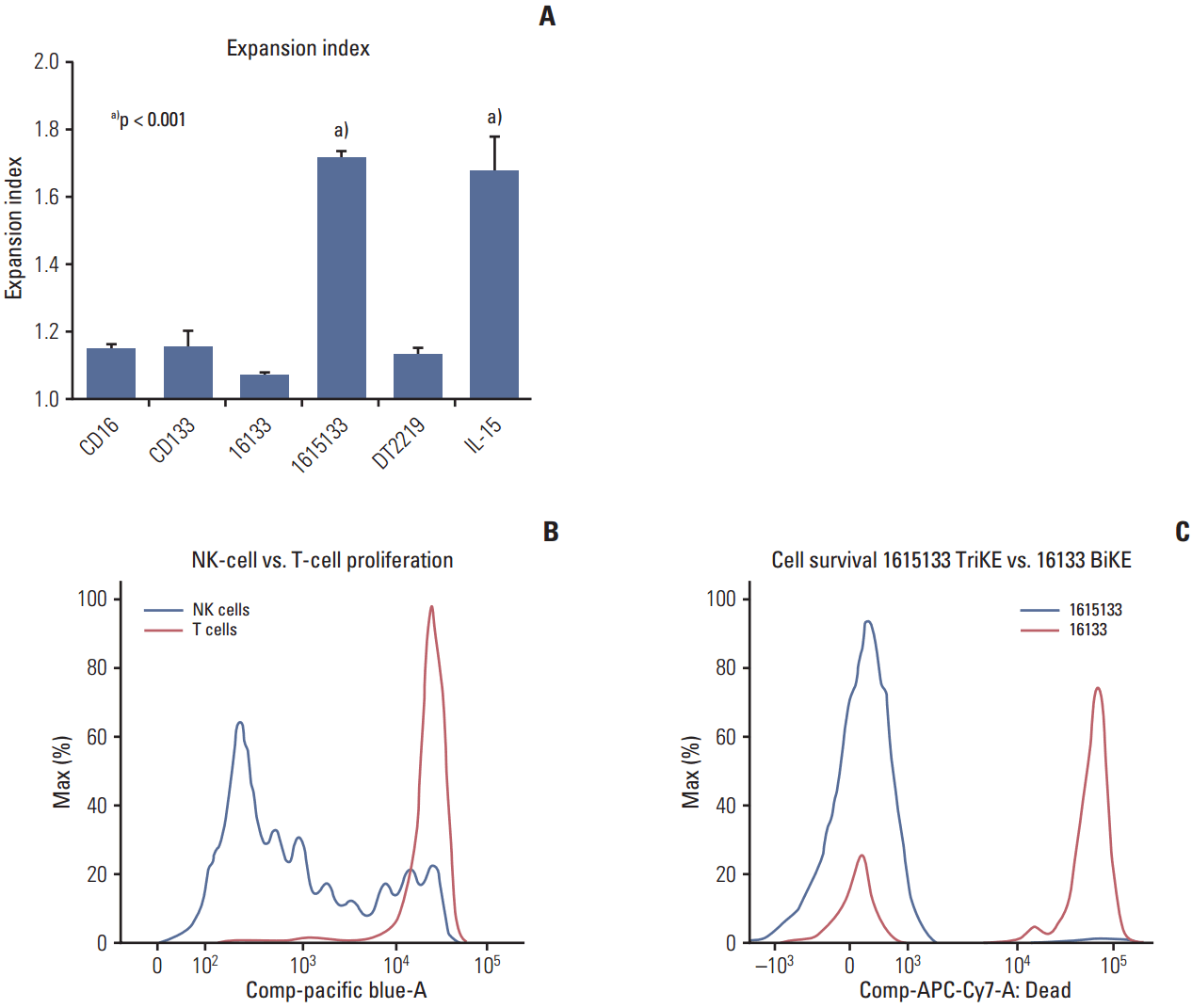
The proliferation induced by the IL-15 moiety of the TriKE was measured by CellTrace dye dilution in viable NK and T cell populations. When the donor PBMCs were exposed to 1615133 TriKE or 16133 BiKE, only the TriKE group induced proliferation (Fig. 2A). Importantly, as shown with previous molecules [14,20], the T cells displayed minimal proliferation after 7 days of drug exposure, suggesting that the expansion was mostly restricted to NK cells (Fig. 2B). A comparison with other control agents, including anti-CD16 scFv, anti-CD133 scFv, DT2219 (a targeted toxin consisting of an anti-CD22 and anti-CD19 scFv linked to a diphtheria toxin), and NCI derived IL-15, showed that only 1615133 TriKE and NCI IL-15 induced proliferation. The other controls showed only the base level proliferation of NK cells (Fig. 2A). To determine the potential of 1615133 TriKE to induce prolonged survival, the purified NK cells were incubated for 7 days with 1615133 or 16133 BiKE. A reactive dye was used to quantify cell death in the different treatment groups. The TriKE group showed a significantly larger amount of live cells, which do not incorporate the reactive dye, compared to BiKE (Fig. 2C). Overall, the results indicate that IL-15 present in 1615133 TriKE induced NK cell proliferation and prolonged their survival.
3. Activity and specificity of lytic degranulation
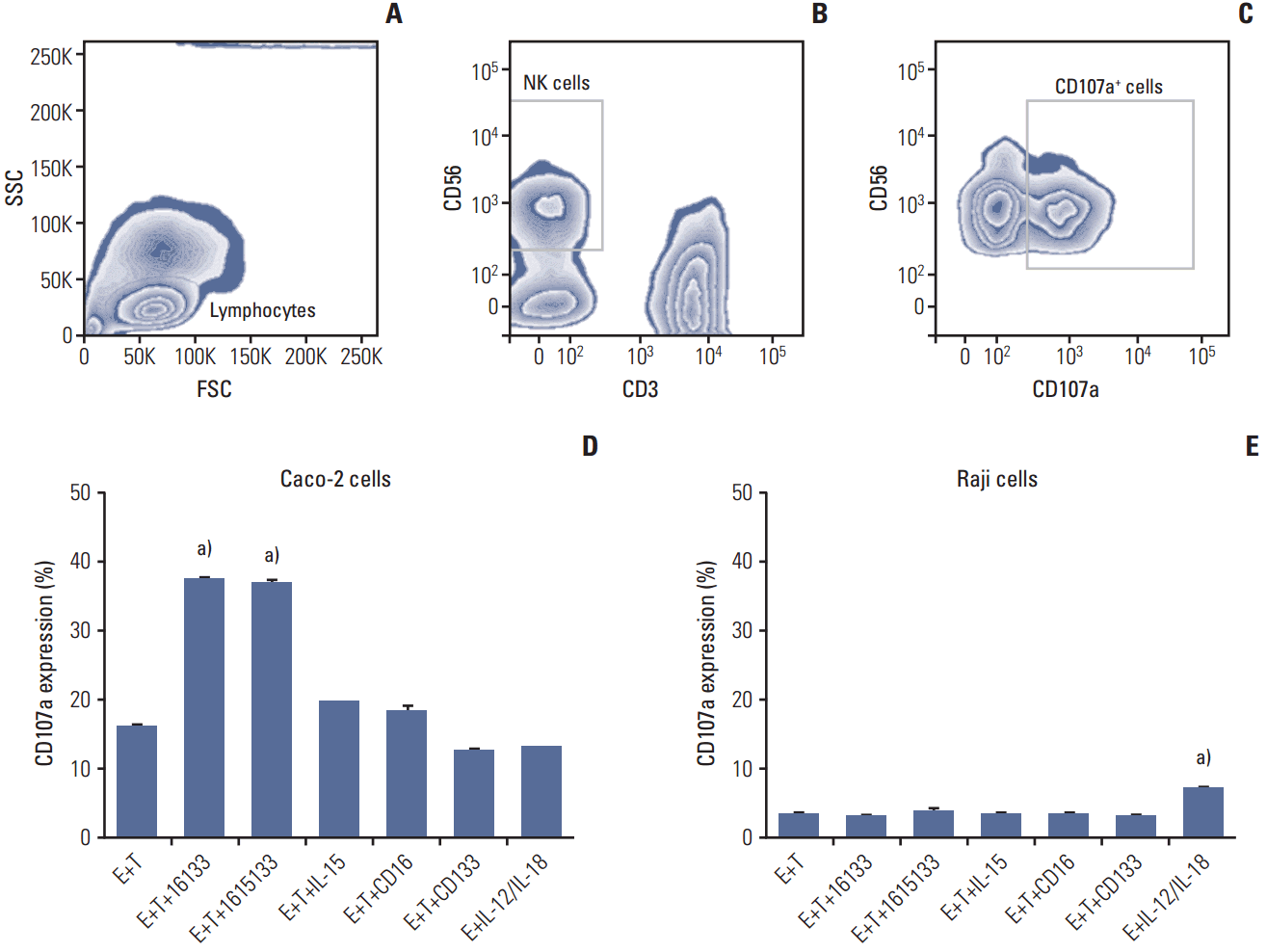
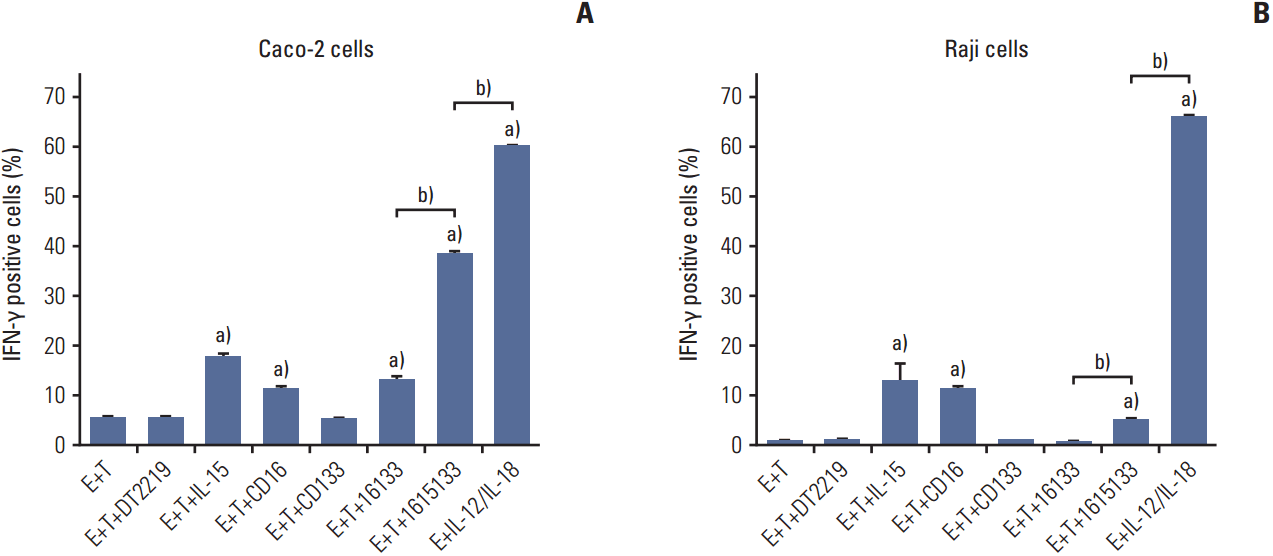
The potential of TriKE to induce lytic degranulation was assessed by examining the induction of CD107a expression on NK cells. As a representative of a CD133 positive cancer population, PBMC were exposed to Caco-2 colorectal carcinoma cells and the coculture was treated with 16133 BiKE, 1615133 TriKE, NCI derived IL-15, anti-CD16 scFv, and anti-CD133 scFv. Representative gating procedure is shown in Fig. 3A-C and expression is estimated in Fig. 3D. BiKE and TriKE showed significantly enhanced NK cell related degranulation compared to the controls (p < 0.001). In Fig. 3E, Raji cells were used as a negative control because they have no CD133 expression. Only supraphysiologic IL-12/IL-18 stimulation, which did not include targets, showed enhanced degranulation here (p < 0.001), but the level did not appear to be biologically relevant. The data indicate that specific NK cell lytic degranulation occurs only in the presence of CD133-expressing targets.
4. Specificity of binding and activity of the TriKE
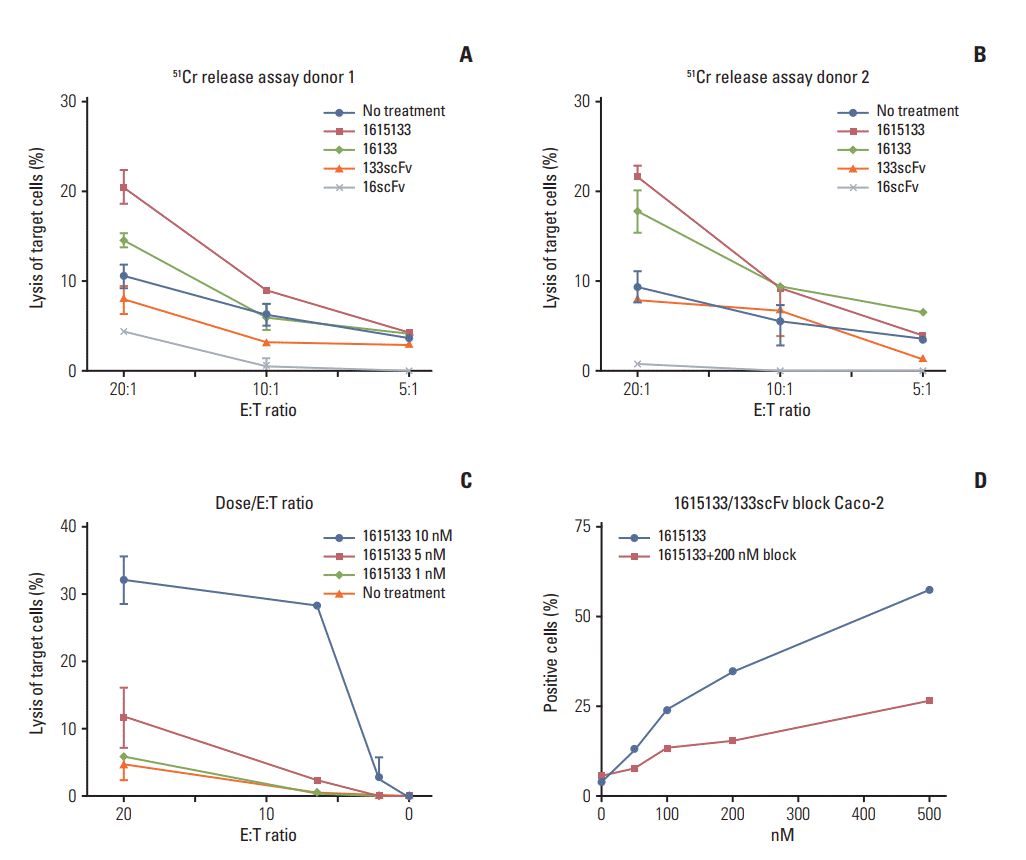
Standard 51Cr release assays were performed to evaluate the functional activity of 1615133 TriKE in the actual killing of tumor targets, rather than just measuring NK cell degranulation. To determine the effects of incorporating IL-15 into the 16133 scaffold, the cytotoxicity was evaluated using NK cells of two separate donors and Caco-2 tumor targets at different E:T ratios (20:1, 10:1, and 5:1) and the differences in activity among 1615133 TriKE, 16133 BiKE, anti-CD16 scFv, anti-CD133 scFv, and no drug treatment were compared (Fig. 4A and B). Owing to baseline variations, the reproducibility was ensured by repeating the assays with different donors. Killing of the Caco-2 targets was elevated in the TriKE samples compared to the controls, but in the CD107a flow cytometric assays, the level of NK cell degranulation was not significantly higher than in the BiKE group. Furthermore, higher NK cell levels, denoted by the higher E:T ratios, showed larger differences in activity with TriKE inducing the most killing. The dose dependent titration of 1615133 TriKE (1, 5, and 10 nM) with a broader spectrum of E:T ratios (20:1, 6.6:1, 2.2:1, 0.7:1, 0.23:1, and 0.08:1) had the highest impact of the drug activity at higher doses (Fig. 4C). To evaluate the specificity of binding, the flow cytometry-based fluorescence intensity was measured after the incubation of Caco-2 cells with FITC labeled 1615133 TriKE at different concentrations (1, 5, 10, 50, 100, 200, and 500 nM). When an unlabeled anti-CD133 scFv was added in addition to 1615133 TriKE, binding was reduced potently, indicating that 1615133 TriKE is binding target to cells specifically through the interaction with CD133 (Fig. 4D). Experiments were also performed with 100 and 500 nM to ensure reproducibility. Together, these results indicate that the ADCC mediated by the TriKE was antigen directed.
5. Ability of IFN-γ induction
To determine if 1615133 TriKE can also induce pro-inflammatory cytokine production in NK cells, flow cytometry was performed to evaluate intracellular IFN-γ production. The PBMCs of healthy donors were exposed to CD133+ Caco-2 targets and 50 nM of DT2219, NCI derived IL-15, anti-CD16 scFv, anti-CD133 scFv, 16133 BiKE, 1615133 TriKE, and supraphysiologic levels of IL-12 and IL-18 (as a positive control). IL-15, anti-CD16 scFv, and BiKE induced a small but significant increase in the proportion of IFN-γ producing cells compared to the effector+target control. On the other hand, 1615133 TriKE induced a significantly higher proportion of cytokine-producing NK cells compared to all tested drugs, with the exception of the IL-12/IL-18 positive control, which displayed the highest level of cytokine producing NK cells (Fig. 5A). The Burkitt lymphoma Raji cell line, which is CD133– was used to evaluate the specificity of IFN-γ induction. IL-15, anti-CD16 scFv, and 1615133 TriKE showed negligible elevated levels compared to the control with effectors +targets alone. TriKE showed significantly elevated IFN-γ levels compared to BiKE, presumably via the IL-15 moiety, but also significantly lower levels than the IL-12 and IL-18 control (Fig. 5B).
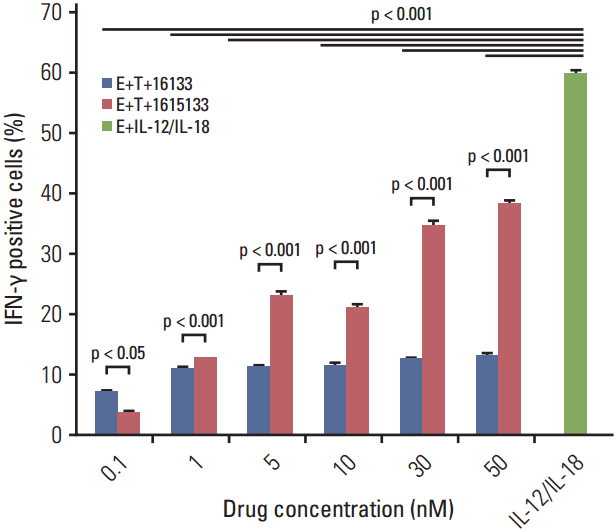
The efficacy of IL-15 in the 16133 scaffold was made more obvious by comparing the dose dependent IFN-γ elevation of 16133 BiKE and 1615133 TriKE. PBMCs were cocultured with CD133+ Caco-2 cells and exposed to the BiKE and TriKE in increasing concentrations (0.1, 1, 5, 10, 30, and 50 nM). Superior NK cell–related IFN-γ production of TriKE was observed at a minimum of 1 nM and increased in a dose dependent manner, whereas the BiKE group showed constant IFN-γ levels that were significantly lower than TriKE. Both drugs showed significantly lower IFN-γ levels than the positive IL-12/IL-18 group (Fig. 6).
Discussion
This study documents the formation and testing of a selfsustaining NK cell engager that targets NK cells and CD133+ cells via anti-CD16 and anti-CD133 scFvs. To improve the mode of action of the previously published 16133 BiKE [19], this platform was used to introduce a modified IL-15 cross linker. The new TriKE mediates the specific and improved ADCC and causes NK cell proliferation and prolonged survival. These characteristics were not inducible either with 16133 BiKE, anti-CD16 scFv alone, anti-CD133 scFv alone, or IL-15 alone. In addition, our group previously demonstrated that this same scFv is capable of targeting CD133+ tumor-initiating cells [19]. We have effectively produced the first CSC-specific NK cell immune engager that is capable of simultaneously mediating ADCC and effector cell expansion that can serve as a sufficient off-the-shelf therapy.
Overall, the data suggest that the 1615133 TriKE could theoretically provide an inexpensive alternative to ongoing CAR T cell approaches. In particular, this molecule possesses characteristics that might better drive the NK cell targeting of CSCs.
The benefits of the combinational use of mABs and cytokines, such as IL-15 and IL-2, to improve NK cell–based immunotherapy have been hypothesized [24]. The safety profile, however, tends to be beneficial with IL-15 considering the clinical trials that used IL-2 in pediatric patients suffering from neuroblastoma and adults with ovarian carcinoma. The authors reported that besides the minor signs of improved anticancer reactivity, IL-2–related toxicity leads to serious adverse events, resulting in dose-limiting administration [25]. Furthermore, a murine model showed that the dose to induce pulmonary vascular leakage was 6 times higher with IL-15 exposure than with IL-2, supporting a superior safety profile of IL-15 [26]. Several clinical studies are ongoing using IL-15 to treat patients with refractory malignant melanoma and metastatic renal cell cancer or as an adjuvant therapy after chemotherapy and lymphocyte transfer in metastatic melanoma. A phase I study included 12 consecutive patients with metastatic melanoma and renal cell cancer, and IL-15 administration showed a response in five patients with a decrease in their marker lesions. In addition to these results, the authors stated the impact in NK cellrelated anti-cancer performance by the mediation of proliferation and prolonged survival [27,28] implying the valuable impact of IL-15 in NK cell related anti-cancer defense. The advantage of introducing the IL-15 molecule to BiKE rather than combining BiKE with IL-15 was observed in previously studied TriKEs 161533 [14] and 1615EpCAM [20]. A side-by-side comparison between the respective TriKEs with BiKEs plus IL-15 showed that the TriKEs induced superior functionality in various entities compared to the separate administration of IL-15. This indicates a different mode of action. Furthermore, targeting via TriKE might improve the accumulation of the IL-15 stimulus at the site of the malignancy. The expectation with 1615133 TriKE is that this molecule will enhance targeting and killing at the site of the CSC pool through the delivery of an ADCC and IL-15 signal.
On the other hand, side effects also occur with IL-15 administration that can cause serious complications. Conlon et al. [27] reported a hemodynamic imbalance and typical cytokine-related complications, such as fever and chills, restricted to Common Terminology Criteria for Adverse Events (CTCAE) grade 3 toxicity. Myelosupressive disturbances were reported in one of twelve patients with the highest CTCAE grade 4 concerning lymphopenia [27].
Owing to the reported short term IL-6, IL-8, and IFN-γ elevations after recombinant human IL-15 administration, this study focused on IFN-γ because it is a well-recognized marker cytokine indicating a cytokine storm. Moderate IFN-γ release after 16133 BiKE exposure has already been recognized [19]. A side-by-side comparison of BiKE and TriKE revealed a dose dependent enhancement of IFN-γ release that was higher with the IL-15–containing drug but moderate compared to the supraphysiologic IL-12/IL-18 control. On the other hand, there is the possibility that by targeting NK cells with IL-15, 1615133 TriKE would restrict IL-15 signaling to that subset alone, possibly decreasing the overall negative impact and adverse effects. Furthermore IFN-γ has antitumor function and is necessary for inducing a productive inflammatory response [29]. Therefore, the moderate IFN-γ release induced by 1615133 TriKE might bolster the anti-cancer effects while reducing the toxicity by targeting IL-15 to NK cells.
The sufficient elimination of CSCs is still an unachieved goal in oncology patients. Characteristics, such as chemoresistance and radio-resistance [1], as well as tumor initiating and self-renewal abilities lead to the persistence of CSCs in the patient and represents a known cause for drug refractory relapse, even after years of remission. Studies describe the occurrence of CSCs in several tumor entities, such as breast, colon, prostate, liver, pancreatic, lung cancer, and head and neck squamous cell carcinoma implying the potential for fundamental improvement in cancer treatment by specifically targeting CSCs. The targeting of CD133+ cells leads to a tumor response in breast, ovarian, and gastrointestinal cancer [30,31] even if CSCs represent only a minor percentage (< 10%) of the entire tumor mass [32]. Similar results have been shown with toxin-containing constructs that sufficiently eliminated CD133+ CSC in vitro as well as in a murine model [30,33]. These findings suggest that the novel 1615133 TriKE, described here, has the potential to curve relapse by targeting the CSC pool.
Note that the authors used a fluorescently labeled anti-CD133 scFv, which is same as the scFv present in 1615133 TriKE, to enrich CD133+ cells via flow cytometric sorting and these cells were used in tumor initiation assays. In an in vivo mouse xenograft model, the enriched cells augmented the size and speed of human head and neck cancer tumor growth [31]. Expedited tumor initiation has been shown to be a hallmark of CSC activity. This data helps validate the targeting of CD133 in TriKE.
CD133 was reported to be expressed in normal stem cells, which are found in the nervous and vascular system as well as in pluripotent stem cells of the hematopoietic system [34-36]. Drug safety analyses in previous studies using a targeted toxin linked to an identical anti-CD133+ scFv (dCD133-KDEL) revealed very little non-target-related toxicity in a murine model and in human in vitro pluripotent stem cell assays [37]. This lack of reactivity to normal stem cells could be attributed to the differential copy number. Even if normal stem cells are being targeted, Rutella et al. [38] described the occurrence of CD133– pluripotent stem cells in bone marrow capable of differentiating into lymphoid, myeloid, and CD133+ cells implicating the sufficient regeneration of the hematopoietic system even after the elimination of CD133+ stem cells [39].
The incorporation of a modified IL-15 crosslinker in the 16133 BiKE scaffold forming 1615133 TriKE shows therapeutic promise. The new TriKE demonstrated an improvement of ADCC. Furthermore, the focused delivery of IL-15 stimulus as part of the same molecule that harnesses NK cells to form an immunological synapse with cancer cells provides prolonged survival and proliferation of effectors. The data suggests that the new construct might address the need to provide a highly active immune engager capable of eliminating CSCs at the root of drug refractory relapse in cancer patients.