Introduction
Genetic and genomic tests are rapidly expanding, and the interpretation of genetic variants is a great challenge now that many genetic variants of unknown significance (VUS) have been identified. Among those genes for which variant interpretation is challenging, BRCA1 and BRCA2 are of particular interest because of the significant clinical implications of pathogenic mutations and the high prevalence of missense VUS that complicate clinical diagnoses and decisions [1].
Germline mutations in BRCA1 or BRCA2 are clinically correlated with hereditary breast and ovarian cancer (HBOC) syndrome, which features early-onset breast cancer, a high incidence of ovarian cancer, and an increased risk of other cancers, including prostate and pancreatic cancers [2]. BRCA1/2 testing is now an essential part of the management of HBOC syndrome, but in practice, approximately 5%-21% of patients tested for BRCA1/2 have one or more VUS, with Asians and African Americans having higher rates [3-7]. Although several public databases, including the Breast Cancer Information Core (BIC, http://research.nhgri.nih.gov/bic/) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), provide estimated risks for certain VUS, interpretation of VUS in Asian populations is still problematic due to limited data.
In response to the need for standardized and intercommunicable assessment, the American College of Medical Genetics and Genomics (ACMG) recently established standards and guidelines for the interpretation of sequence variants [8]. The standards and guidelines suggest a 5-tier variant classification with the following categories: pathogenic (P), likely pathogenic (LP), VUS, likely benign (LB), and benign (B). These classifications are determined by incorporating evidence from population, computational, functional, familial segregation, de novo, allelic, and database data. Nevertheless, there may still be issues of ambiguity and subjectivity in some analyses; therefore, applying objective criteria will be important to increasing reliability and reducing the uncertainty associated with the interpretation. Comparing variant frequencies in the general population is a good example of such an objective measure that has become an essential part of the ACMG guidelines. The ACMG recommends that the odds ratio (OR) be calculated based on the occurrence of the variant in cases and in the general population. Additionally, the lower limit of a 95% confidence interval (CI) of an OR that exceeds 1.0 should be regarded as strong evidence of pathogenicity (PS4) [8]. However, there is still debate regarding which population data should be used for OR calculations.
Recent studies have attempted to reclassify variants according to the ACMG guidelines [9-12]; however, the proportion of VUS is still high, particularly in patients from ethnic groups for which there is insufficient genetic and genomic information [12]. Here, we evaluated BRCA1/2 variants in Korean patients suspected of having HBOC according to the ACMG guidelines. Our simulation demonstrated that the optimal number of accurate ethnic controls was needed for rare pathogenic variants to be properly scored by this statistical approach.
Materials and Methods
1. Subjects
This study included 745 women with at least one clinical feature of HBOC who were evaluated for BRCA1/2 germline mutational status at Yonsei Cancer Center between January 2008 and January 2016. The clinical features of HBOC included (1) at least one first- or second-degree relative with breast and/or ovarian cancer, (2) breast cancer diagnosed before age 40, (3) bilateral breast cancer, (4) breast cancer and synchronous or metachronous ovarian cancer, and (5) epithelial ovarian cancer. We observed breast tumors in 596 patients, ovary tumors in 124 patients, and both breast and ovary tumors in 25 patients. The World Medical Association Declaration of Helsinki on medical research protocols and ethics was followed throughout the study. The institutional review board reviewed and approved this study.
2. BRCA1 and BRCA2 testing and variant analysis
After extraction of patient genomic DNA from peripheral blood, the entire coding regions and exon-intron boundaries (±20 base pairs) of the BRCA1 and BRCA2 genes were analyzed by Sanger sequencing. Reference sequences used for analysis were NM_007294.3 for BRCA1 and NM_000059.3 for BRCA2. Variant descriptions followed the nomenclature system of the Human Genome Variation Society (http://www.hgvs.org/mutnomen). Variants were originally reported by a 3-tier system, and those reported as disease-causing mutations or VUS were further evaluated and reclassified according to the ACMG guidelines as described below.
3. Statistical analysis using population control data
Various population data were used for the statistical analysis of each variant. From the Exome Aggregation Consortium (ExAC), allele frequencies in 52,758 individuals, excluding those who belong to the Cancer Genome Atlas consortium, were retrieved. For each variant site, genotype data passing the quality cutoffs as defined by the ExAC were analyzed. The ExAC populations used for final analysis included: 4,533 African/African American (AFR), 5,608 Latino (AMR), 3,933 East Asian (EAS), 3,307 Finnish, 27,173 Non-Finnish European (NFE), and 8,204 South Asian individuals. We also retrieved allele frequencies from 1,314 Koreans, including 622 individuals from the Korean Reference Genome DB (KRGDB, http://152.99.75.168/KRGDB/) and 692 individuals from the database of a commercial laboratory, the Theragen Etex Bio Institute (Suwon, Korea). For each variant, the OR was calculated based on its occurrence in 745 patient cases and in population controls described above. For those variants that were not present in the population controls, the corrected OR was calculated by adding 0.5 to all cells in a 2×2 table, as previously described [13].
4. Other assessments for ACMG classification
Computational predictions for missense variations were calculated using the SIFT, PolyPhen-2, MutationTaster, MutationAssessor, and FATHMM algorithms [14]. Pathogenic or benign evidence was scored when predictions of all algorithms agreed. GVGD prior probability was also retrieved (http://agvgd.iarc.fr/) for comparison. Splice site analysis was determined using the SpliceFinder-like, MaxEntScan, NNSPLICE, GeneSplicer, and Human Splicing Finder algorithms implemented in the Alamut Visual software (Interactive Biosoftware, Rouen, France). Although family member genotypic information was unavailable for most VUS, familial segregation analysis was performed wherever possible using a recently described method [15]. By reviewing all available disease and literature databases, including the BIC, ClinVar, Sharing Clinical Reports Project (SCRP), PubMed, and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/), functional and database data were evaluated and utilized when sufficient information was available.
5. Structural modeling
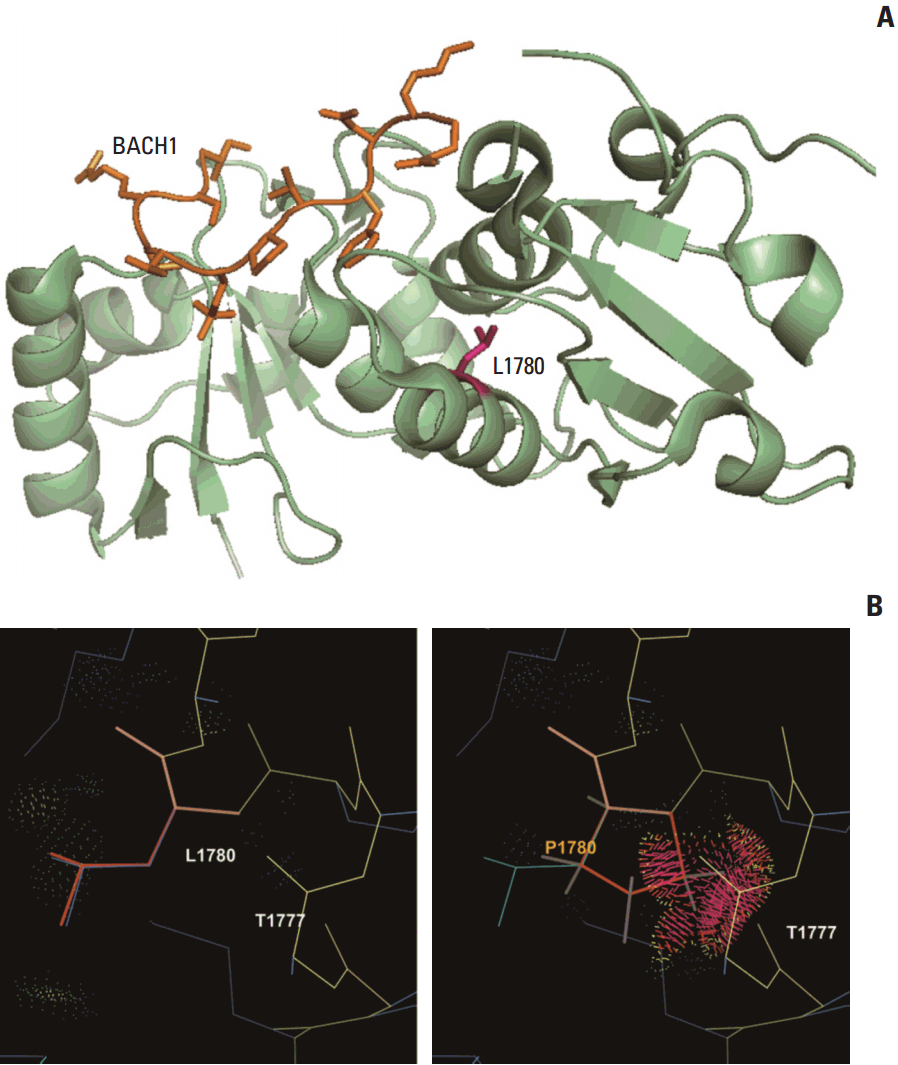
To evaluate the structural impact of the BRCA p.Leu1780Pro mutation, a crystal structure of the BRCA1 BRCT repeats bound to a phosphorylated BACH1 peptide (1T29) was retrieved from the RCSB Protein Data Bank, and the location of the variant was modeled with the PyMOL software (Schrödinger, New York, NY). Sidechain rotamer analysis was performed using the KiNG software (Kinemage, Next Generation) [16].
6. Simulation of 95% CIs of ORs in cases and controls
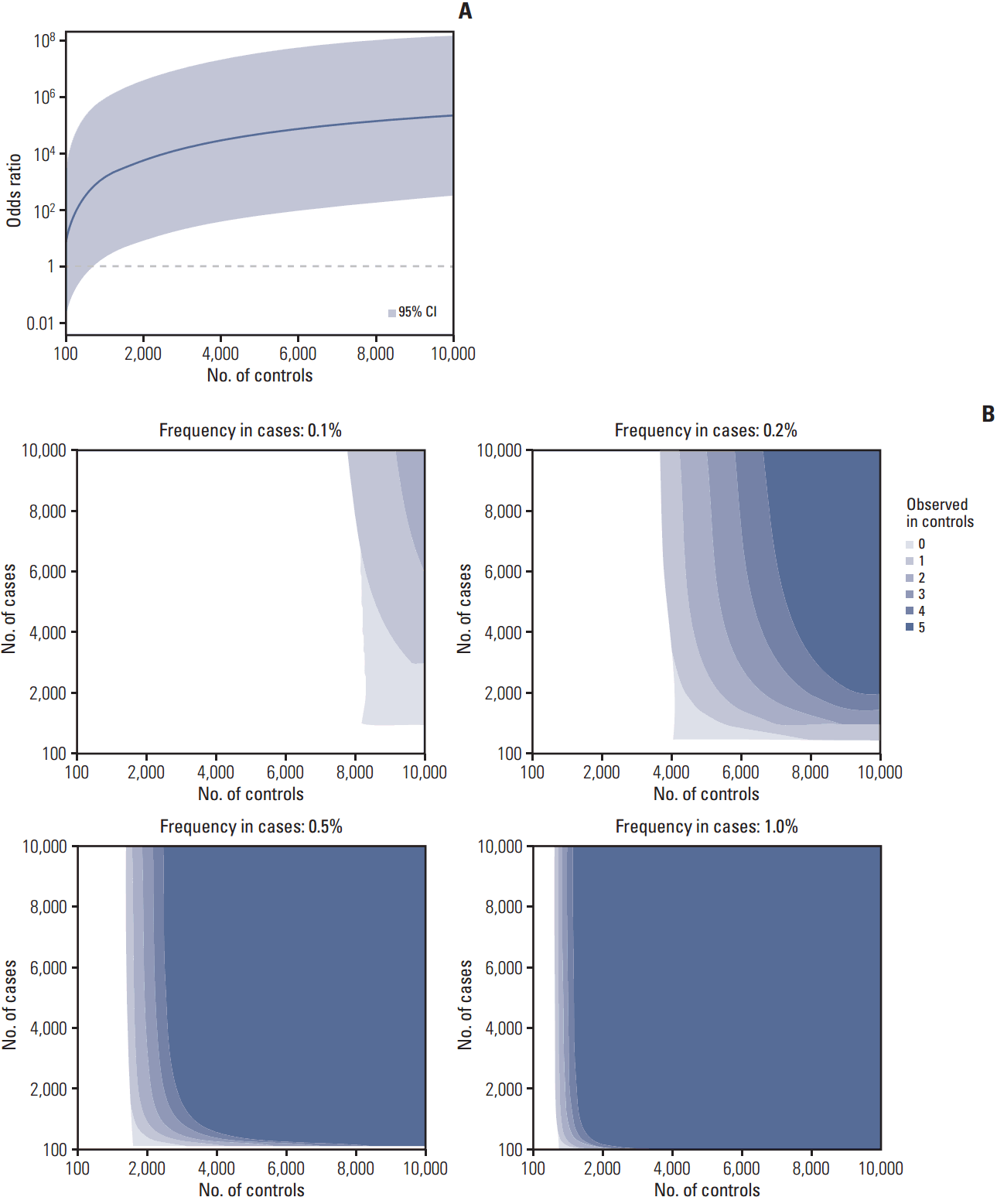
To estimate adequate numbers needed for statistical analysis, we calculated the OR and 95% CI by simulating the total number of cases and controls from 100 to 10,000, a variant frequency in cases from 0.1% to 10%, and the number of control individuals carrying the same variant from 0 to 5.
Results
1. Characterization of variants for reclassification
We revisited 144 variants (69 variants of BRCA1 and 75 variants of BRCA2), including 58 variants reported to be disease-causing mutations and 86 reported to be VUS in the original 3-tier classification. All disease-causing mutations were null variants and included 24 nonsense, 26 frameshift, and eight splice-site mutations. The VUS included 64 missense, 14 synonymous, and eight intronic variants. Furthermore, we compared our cohort to an additional cohort of 715 Korean patients with breast cancer [12] that included 23 missense VUS.
2. Population data analysis
We classified two BRCA2 variations, p.Ile3412Val and p.Ile2490Thr, to meet benign criterion weighted as standalone (BA1) because the allele frequencies exceeded 5% in ExAC, AFR, and AMR populations. Because the incidence of HBOC is estimated to be between 1 in 400 and 1 in 800 [17], we defined a minor allele frequency (MAF) > 0.1% as too high for the disorder, and therefore classified it as BS1 (benign criterion weighted as strong-1; allele frequency is greater than expected for disorder). This is still a conservative measure considering that many different BRCA1/2 mutations have been reported, most of which are not highly prevalent mutations except for those occurring within the Ashkenazi Jewish population [17]. Using MAFs calculated from six different ExAC populations, we could classify 27 variants as BS1. Using MAFs from 1,314 Korean exomes, we identified an additional nine out of 36 variants that we classified as BS1.
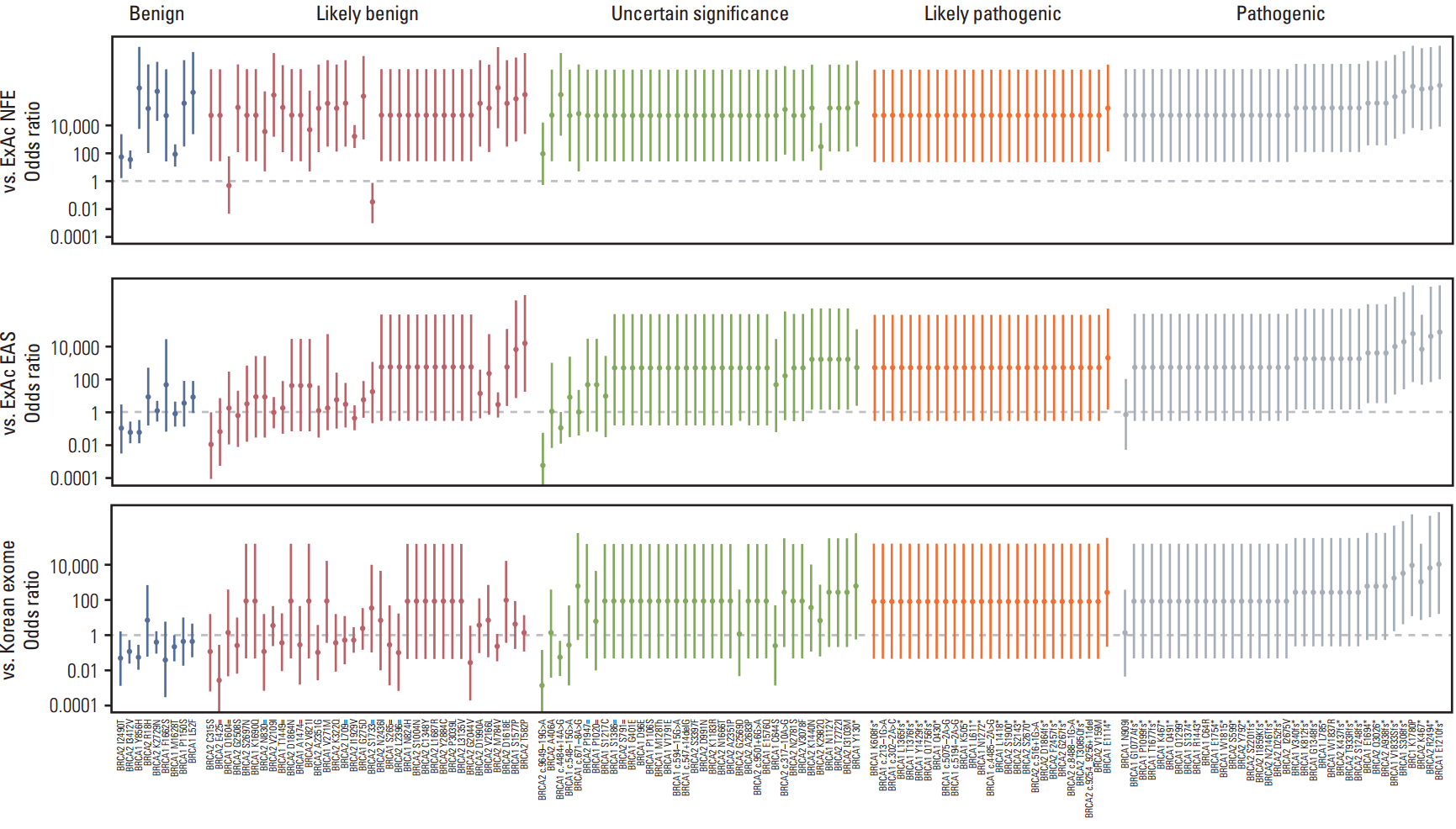
We found that ORs were falsely elevated when we used ExAC data for control populations. For example, use of the ExAC NFE data resulted in the lower limits of the 95% CI being 1.0 for most variants (Fig. 1, S1 Table). Even when the ExAC EAS data was used, the ORs of some variants, including BRCA1 p.Ser1577Pro, BRCA2 p.Thr582Pro, and BRCA2 p.Asp1618Glu, were overinflated. Using Korean data, the false positives could be excluded, and six variants could then be classified as PS4 (pathogenic criterion weighted as strong-4; the prevalence of the variant in affected individuals compared with the prevalence in controls). Interestingly, among this group, we identified a missense variant, BRCA1 c.5339T->C (p.Leu1780Pro; rs80357474), which was a previously reported VUS. However, in our analysis, this variant had a significantly high OR (41.2; 95% CI, 2.4 to 699.5). Apart from these six variants, 88 (including 52 null variants) were absent from the population database and were classified as PM2 (pathogenic criterion weighted as moderate-2; absent from controls).
3. Computational, functional, familial, and database data analysis
Among 64 missense variants, 18 were predicted to have no effect on the gene product according to all five computational algorithms and were classified as BP4 (benign criterion weighted as supporting-4; multiple lines of computational evidence suggest no impact on gene or gene product). In contrast, 13 variants were predicted as deleterious using all algorithms and were classified as PP3 (pathogenic criterion weighted as supporting-3; multiple lines of computational evidence support a deleterious effect on gene or gene product). None of the 14 synonymous variants were predicted to affect splicing and were classified as BP7 (benign criterion weighted as supporting-7; a synonymous variant for which splicing prediction algorithms predict no impact on the splice consensus sequence nor the creation of a new splice site and the nucleotide is not highly conserved). Seven missense variants were reported as deleterious by previous functional studies and classified here as PS3 (pathogenic criterion weighted as strong-3; well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product). Familial segregation and de novo data could not be obtained for most patients. None of the investigated families fulfilled the criteria for evidence [15] due to the small number of family members who were genotyped.
4. ACMG classification
All of the 58 null variants that were previously classified as disease-causing mutations were classified as P or LP, except for BRCA1 p.Tyr130Ter (rs80356888), which was present in one individual among 3,933 ExAC EAS controls and therefore could not be classified as PM2. Six among 86 variants (7.0%) previously reported as VUS were reclassified as P or LP, 45 (52.3%) were reclassified into B or LB, and the remaining 35 (40.7%) remained classified as VUS (S2 Table). Among the seven high-frequency VUS (> 1% of all patients), only BRCA1 p.Leu1780Pro was classified as P, while the remaining ones were classified as B or BP (S3 Table). In total, 126 of the 745 patients (16.9%) with features of HBOC harbored variants (63 types) that were classified as P or LP according to the ACMG classification.
5. BRCA1 p.Leu1780Pro is a founder mutation in Koreans
BRCA1 p.Leu1780Pro, a missense VUS remarkably enriched in patients over population controls, was classified as P using multiple evidence categories, including PS4, PS3, and PP3. This variant was observed in 11 of our 745 patients (1.5%), which was equal to 8.7% of the 126 patients with P or LP variants. All five algorithms predicted this variant as deleterious. Leu1780 is located at the C-terminal end of the BRCT domain, an area in which other pathogenic missense variants have been reported [18,19], but it does not appear to be directly involved in substrate binding. The replacement of leucine with proline, which has a bulky aromatic side chain, is predicted to cause a side-chain clash with adjacent amino acids and disrupt the α-helical structure of the BRCT domain (Fig. 2).
The clinicopathological features of 11 patients with the BRCA1 p.Leu1780Pro mutation are presented in Table 1. Among them, 10 were diagnosed with breast cancer, and the median age at the time of first diagnosis was 37 (range, 34 to 53). Most of these cases (8 of 10) were triple-negative breast cancer. Three patients were diagnosed with epithelial ovarian cancer, two of which had both breast cancer and ovarian cancer. Additionally, two patients had bilateral breast cancers. Six of the 11 patients had at least one first-degree relative with breast cancer and/or ovarian cancer (Table 1, S4 Fig.). Although these findings were very typical of HBOC, we did not classify them as PP4 because of the locus heterogeneity that exists in breast and ovarian cancers [9]. Finally, patients carrying the BRCA1 p.Leu1780Pro mutation lacked any other pathogenic BRCA1 or BRCA2 mutations.
6. Simulation to determine the optimal numbers of subjects for OR calculations
Because we could effectively obtain evidence and discover pathogenic variants using statistical approaches, we explored how many cases and controls would be needed to reach statistical significance. For a variant, increasing the number of controls can increase the OR and statistical significance, and the minimum number of controls needed varies depending on the frequency of the variant in cases and in controls. If a variant is present at a frequency of 1% (10 in 1,000 cases) and absent from controls, at least 820 controls are needed for the lower limit of a 95% CI to exceed 1.0 (Fig. 3A). Moreover, although more than 4,000 controls are needed for a variant existing in 0.2% of cases and 0% of controls, 500 cases are sufficient for a statistically valuable calculation, and increasing the number of cases provides little additional advantage. However, more cases and controls are needed if one or more individuals with the variant are present in control populations (Fig. 2B). For variants existing at a high proportion in cases (e.g., > 1%), small numbers of cases and controls are sufficient to reach statistical significance (S5 Fig.).
Discussion
The ACMG guidelines were developed to improve and standardize classification of potentially pathogenic genetic variants by defining 28 criteria. Nevertheless, ambiguity still exists in applying certain criteria; therefore, efforts are ongoing to increase the clarity of criteria and improve concordance in variant interpretation among different laboratories and geneticists [9]. Using general population data may be one of the most powerful tools for selecting pathogenic variants and excluding benign polymorphisms in an objective manner. However, our results show that caution should be exercised when population data from different racial or ethnic groups are used in analyses. Korea is located between China and Japan; therefore, the genetic characteristics of Koreans are considered to be similar to those of Chinese and Japanese people. However, when we used the ExAC EAS data in our analysis, which is mainly composed of Chinese and Japanese individuals, we calculated misleadingly high ORs for some variants. Because there are likely Korean-specific polymorphisms, using relevant Korean population controls is the best way to avoid inaccurate conclusions.
Additionally, our simulation raises issues and concerns regarding the optimal number of population controls. This varies depending on the frequency of the variant in cases. For rare pathogenic variants, a large number of controls is needed. For example, the BRCA1 p.Leu1780Pro variant that was present in 1.5% of cases, was marginally significant (OR, 19.5; 95% CI, 1.1 to 331.5) when 622 Korean controls from KRGDB were used. Considering that p.Leu1780Pro was the second most prevalent mutation in our series, it is likely that tens of thousands of relevant controls are needed for most other rare variants.
Classifying VUS into pathogenic or neutral variants is a challenge for clinicians evaluating the BRCA1 or BRCA2 mutation status of patients with features of HBOC. Efforts are being made to reclassify VUS and reduce the rate at which they are reported. For example, Myriad Genetic Laboratories reduced the VUS rate to 2.1% using their exclusive database [20,21]. Open-access databases and research consortia, including ClinVar, the BIC and the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) also play important roles in the interpretation of VUS. Nonetheless, these databases primarily consist of patients from Western countries, making it difficult to use these populations to interpret the clinical significance of variants found in Asian or other ethnic populations.
The BRCA1 p.Leu1780Pro variant appears to be specific to Koreans. Only one Asian case (detailed ethnicity unknown) with BRCA1 p.Leu1780Pro was listed in the BIC, and this was submitted by Myriad Genetics Laboratories and graded as VUS. ClinVar and SCRP also list three submissions with a VUS interpretation. Interestingly, other reports of this variant are restricted to studies using Korean cohorts, all of which classify the variant as VUS [12,22-24]. In our study, this variant was found in 11 cases and was the second most common mutation after BRCA1 c.3627dupA (12 cases). BRCA1 Leu1780 is located at the α1-helix of the C-terminal end of two BRCT domains (amino acids 1,646-1,859), which are critical for DNA repair activity. In response to DNA damage, the BRCT domain binds to phosphorylated proteins that are essential for the DNA damage response [25]. Pathogenic missense variants reported to date have mainly been located at the BRCT domain [18,19]. Lee et al. [26] conducted a comprehensive functional analysis of missense variants in the BRCT domain using multiple assays, including proteolysis tests, to measure protein folding stability, phosphopeptide binding assays to measure binding activity and specificity of peptide interaction, and transcriptional assays to measure transcriptional activation activity. They found that the BRCA1 p.Leu1780Pro mutation causes adverse effects on cells in the above assays.
We observed that the clinicopathological features of patients carrying BRCA1 p.Leu1780Pro were typical of HBOC (S6 Table) [17]. Although all enrolled cases were suspected HBOC cases, the clinical features of patients with BRCA1 p.Leu1780Pro were remarkable among the enrolled subjects. For familial investigation, information regarding the parents’ history and genotype was unavailable for most families, in part because of the influence of the Korean War, followed by the division of territory, and infrequent visits to clinics until the mid-20th century. Issues related to incomplete penetrance over a lifetime, inclusion of pathogenic variants in the general population controls, and missense variants with hypomorphic characteristics associated with moderate to low risk likely remain [27].
In conclusion, we applied the ACMG guidelines and reclassified variants in BRCA1/2 in Korean patients with features of HBOC. We found that East Asian control populations are inappropriate for accurate OR calculations, and the use of relevant Korean controls helped us to identify pathogenic variants, including one founder mutation, BRCA1 p.Leu1780Pro. Our simulation demonstrated that a sufficient number of controls is needed for rare variants to be statistically evaluated (for instance, more than 820 relevant ethnic controls for a variant existing in 1% of cases); therefore, large genome projects for individual ethnic groups will be needed in the future for more accurate interpretation of genetic variants.