Introduction
The molecularly-targeted drugs that inhibit epidermal growth factor receptor (EGFR), gefitinib and erlotinib, were initially expected to treat the vast majority of non-small cell lung cancer (NSCLC) because overexpression of EGFR protein is frequently detected in this type of tumor [1]. However, many preclinical and clinical studies have demonstrated that these drugs are only effective in a specific subset of NSCLC, in which the mutations are carried on the kinase domain of the EGFR gene [2,3]. Thus, EGFR tyrosine kinase inhibitors (TKI) are a first-line therapy in patients with advanced NSCLC with EGFR mutations based on several randomized phase III studies showing a significantly improved response rate (RR) of about 70% and median progression-free survival (PFS) of about 10 months [4-8]. However, only 10% of NSCLC patients in the United States and 35% in East Asia have tumors with EGFR mutations [2,3]. Patients with wild type (WT) EGFR, who constitute a larger proportion of NSCLC patients, derive limited clinical benefits from EGFR-TKIs. In a meta-analysis study analyzing 11 randomized controlled studies, including 1,605 patients with advanced WT EGFR NSCLC, first-generation EGFR-TKIs showed a significantly lower RR of 7.2% and a higher risk of progression of 1.41 when compared with conventional chemotherapy in patients with WT EGFR NSCLC [9]. Although one EGFR-TKI, erlotinib, was approved in patients with WT EGFR NSCLC, the survival benefit was modest in the BR.21 trial comparing this drug with a placebo in an unselected pretreated NSCLC population. Specifically, the improvement in median PFS was 0.4 months and the improvement in median overall survival (OS) was 2.0 months [10]. Thus, there are major unmet needs in using EGFR-TKIs to treat patients with advanced NSCLC with WT EGFR.
We previously conducted a randomized phase II clinical study to improve gefitinib efficacy by adding simvastatin in unselected advanced NSCLC patients [11]. One of the main reasons for primary resistance to EGFR-TKIs is that there are alternative mechanisms for persistent activation of EGFR downstream signaling, including both the RAS/ERK and phosphoinositide 3-kinase (PI3K)/Akt kinase pathways [12]. Thus, we hypothesized that simultaneous inhibition of both pathways would inhibit tumor cell survival more effectively in tumors resistant to EGFR-TKIs. One candidate drug combination is EGFR-TKIs and statins, which are irreversible inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase that are used to treat hypercholesterolemia by blocking the mevalonate biosynthesis pathway [13]. Isoprenoids, which are products of the mevalonate pathway, are attached to RAS proteins to allow them to anchor in the cell membrane, where they perform biological roles [14,15]. By interrupting the biosynthesis of mevalonate, statins may inhibit activation of RAS and downstream signaling cascades, including the RAF/MEK/ERK and PI3K/AKT cascades, which play critical roles in regulating cell survival and proliferation. Thus, this may be a promising therapeutic approach to overcoming tumor resistance to EGFR-TKIs. In our previous study, the combination of gefitinib and simvastatin showed higher RR (39% vs. 8%, p=0.06) than gefitinib alone in the subgroup of nonadenocarcinomatous (NA)-NSCLC, which is unlikely to have EGFR mutations. This finding suggests that simvastatin may enhance sensitivity to gefitinib in NA-NSCLC that is resistant to gefitinib. Moreover, several preclinical studies demonstrated that combined gefitinib and lovastatin exerted significant synergic cytotoxic effects in vitro in squamous cell carcinomas, NSCLC, and colon carcinoma cell lines that do not possess the activating EGFR mutations [16,17].
Simvastatin is metabolized in liver cells by CYP3A4, and its concurrent use with other substrates of this enzyme is contraindicated because of increased toxicity [18]. Thus, the combination of simvastatin and gefitinib or erlotinib would not be suitable for a clinical study because they are also CYP3A4 substrates [19]. However, afatinib, a second-generation irreversible EGFR-TKI that is not a CYP3A4 substrate, was identified as a good candidate for combination with simvastatin. Therefore, we conducted a randomized phase II study to compare the efficacy of afatinib and simvastatin (AS) with afatinib alone (A) in previously treated patients with NA-NSCLC.
Materials and Methods
1. Patients
Eligible patients were ≥ 18 years old with pathologically confirmed stage IIIB/IV NA-NSCLC (e.g., squamous cell or large cell carcinoma) that progressed after first- or second line cytotoxic chemotherapy regimens, including at least one platinum-containing regimen. Patients with an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0, 1, and 2 and adequate organ function were eligible. Patients had a measurable lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST) ver. 1.1 [20]. Patients receiving prior treatment with small molecules or antibodies that inhibit EGFR (e.g., gefitinib, erlotinib, and cetuximab) were excluded.
All patients provided written informed consent, and this study was approved by the Institutional Review Board. The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization/Good Clinical Practice. This study is registered with ClinicalTrials.gov under identifier NCT01156545.
2. Study design and treatment
This was a multicenter, open, randomized, phase II study to evaluate the synergistic effects of afatinib plus simvastatin in pretreated NA-NSCLC patients. The stratification factors for randomization were ECOG PS (0 vs. 1 or 2).
After allocation to the treatment arms, patients received continuous daily treatment with either afatinib plus simvastatin or afatinib plus the best supportive care until disease progression, unacceptable adverse events, or another reason necessitating withdrawal. The treatments were administered as 28-day courses. The starting doses of AS were 40 mg once daily. Dose escalation of afatinib to 50 mg was permitted for individual patients after one cycle if there were minimal adverse events at 40 mg. When drug-related adverse events occurred, the afatinib dose was reduced in increments of 10 mg, with the lowest dose being 20 mg. There was no dose reduction for simvastatin.
3. Tumor assessment
The tumor response was assessed after 4, 8, 16, and 24 weeks and every 8 weeks thereafter until progression or withdrawal for another reason. After week 48, the response was assessed every 12 weeks. Tumor response and progression were assessed using RECIST ver. 1.1 [20].
4. Statistical analyses
The primary endpoint was the objective response, as determined according to RECIST ver. 1.1 [20]. The objective response rate (ORR) was calculated as the ratio of the number of responders to the number of patients assessed for tumor response. Responders were defined as patients showing complete response (CR) or partial response (PR). We expected that afatinib plus simvastatin would increase ORR by 35% from 10% for afatinib alone based on the results of Han et al. [11]. Including 62 patients (31 in each arm) would give statistical power of 80% by Fisher exact test (one-sided, α=0.15). Considering a 10% attrition rate, we planned to recruit a total of 68 patients (34 in each arm).
The secondary endpoints were PFS, OS, and adverse events. Adverse events were graded according to the Common Terminology Criteria for Adverse Events ver. 4.0. PFS was calculated as the duration from the date of randomization to the date of disease progression or death, whichever occurred first. OS was calculated as the duration from the date of randomization to the date of death. Survival time was estimated using the Kaplan-Meier method, and the survival difference between groups was assessed using the log-rank test. p-values were one-sided. A p < 0.025 was considered to be significant.
5. Exploratory biomarker study
Archival tumor tissues were collected before treatment in all patients. We checked the EGFR status by three methods: mutation status by direct sequencing, gene copy number by fluorescence in situ hybridization (FISH), and protein expression by immunohistochemistry (IHC) staining.
Nucleotide sequencing of the kinase domain of the EGFR gene (exons 19, 20, and 21) was performed by direct sequencing of the individual exons. The details describing the sequencing procedure have been described elsewhere [21]. The EGFR gene copy number was calculated with the FISH test using the LSI EGFR SpectrumOrange/CEP 7 Spectrum-Green Probe (Vysis, Abbott Molecular, Des Plaines, IL) according to the standard protocol [22]. Tumors were considered to have an increased copy number (FISH-positive) if they showed gene amplification or chromosome 7 high polysomy based on previously described criteria [22]. The intensity of EGFR IHC staining (Zymed, Thermo Fisher Scientific, Fremont, CA) was scored as follows: 0, no membrane staining; 1+, faint, partial membrane staining; 2+, weak, complete membrane staining in > 10% of tumor cells; 3+, intense complete membrane staining in > 10% of tumor cells. Those with a score of 2+ or 3+ were classified as IHC-positive.
Results
1. Patient characteristics
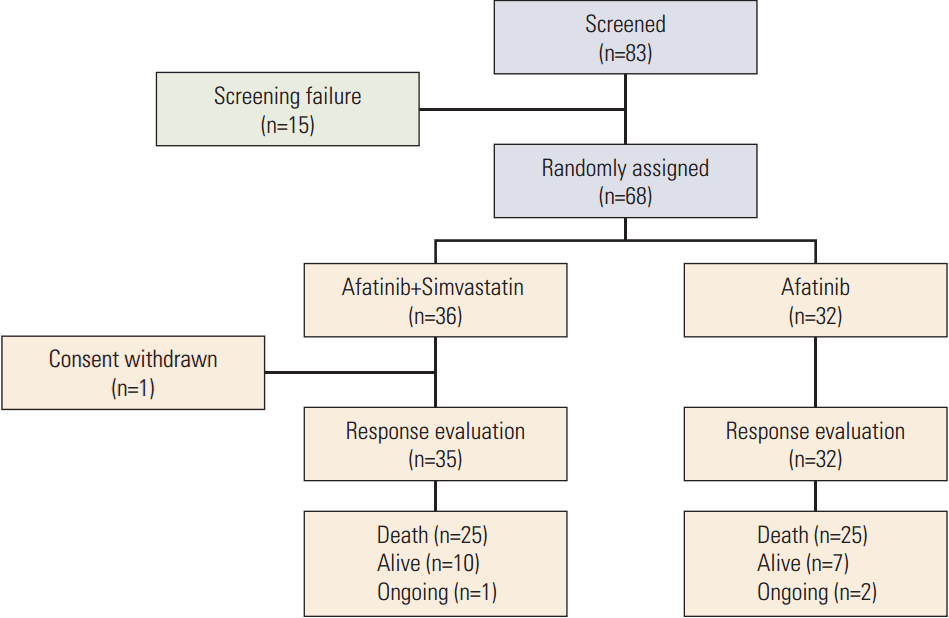
Between November 2012 and September 2015, 83 patients from two centers in Korea were screened and 68 patients were enrolled (Fig. 1). Thirty-six patients were allocated to the AS arm and 32 to the A arm. After stating the allocated treatment, six patients discontinued the study due to toxicity (n=1) or patient refusal (n=5) (one in the AS arm and five in the A arm). Patient characteristics are listed in Table 1. Most patients were male (87%), ever-smokers (92%), had squamous cell carcinoma histology (94%), and good ECOG 0-1 performance (75%). Other histological subtypes included sarcomatoid carcinoma (n=1) and NSCLC-not otherwise specified (n=3). Demographic baseline characteristics, except age, were generally balanced between the two groups.
2. Efficacy
Sixty-seven patients were assessed for response evaluation. One patient showed a CR and four patients showed a PR. The ORR was 5.7% (95% confidence interval [CI], 0.7 to 18.7) for the AS arm and 9.4% (95% CI, 2.0 to 25.0) for the A arm (p=0.430) (Table 2).
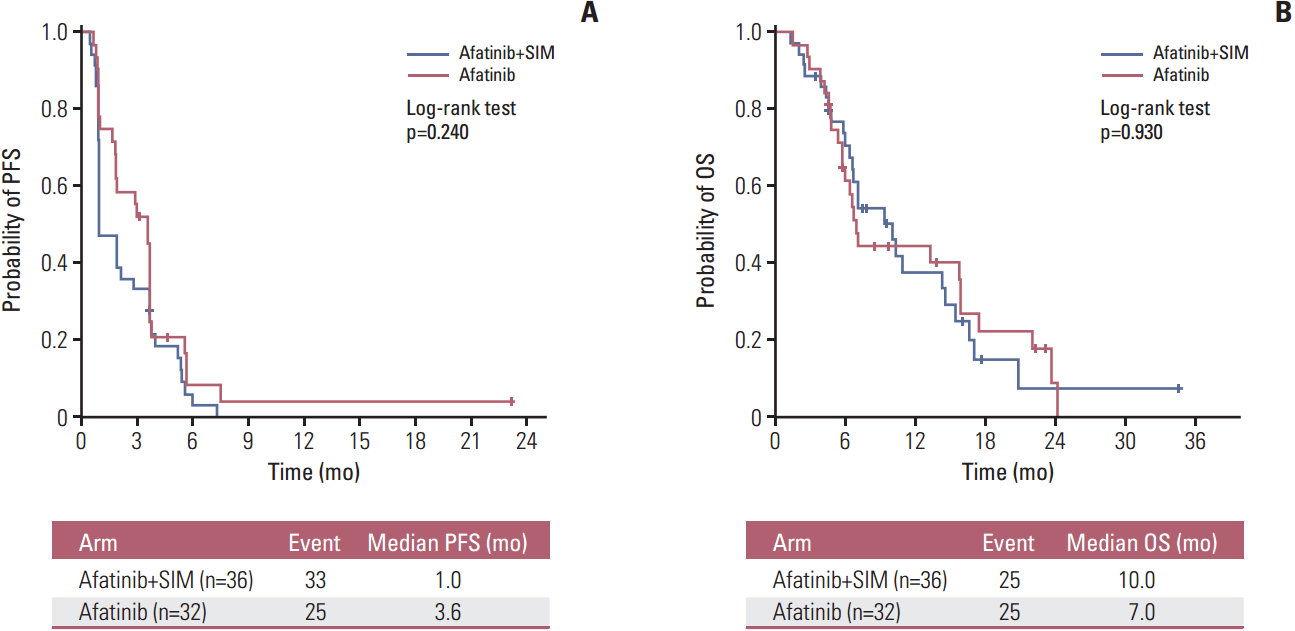
The median follow-up time for all patients was 22.3 months. A total of 50 deaths (73.5%) occurred during data analysis. There was no difference in median PFS between the AS group (1.0 month [95% CI, 0.5 to 1.4]) and the A group (3.6 months [95% CI, 3.0 to 4.1]) (p=0.240) (Fig. 2A). The hazard ratio (HR) for progression was 1.38 (95% CI, 0.84 to 2.29; p=0.898). The median OS was 10.0 months (95% CI, 6.4 to 13.8) for the AS arm and 7.0 months (95% CI, 6.1 to 7.9) for the A arm (p=0.930) (Fig. 2B). The HR for death was 1.03 (95% CI, 0.58 to 1.80; p=0.466). Subsequent chemotherapy after progression was given to 27 of the 35 patients (77.1%) in the AS arm and 18 of the 28 patients (64.3%) in the A arm (p=0.262).
3. Safety
No patient in either arm received an escalated dose of afatinib of 50 mg/day. The dose of afatinib was reduced to 30 mg/day in 14 patients (20.6%) (six in the AS arm and eight in the A arm), whereas the dose was reduced to 20 mg/day in one patient in the AS arm. The main reasons for the dose reduction were stomatitis and skin rash. Five patients (7.4%) stopped treatment because of adverse events (two in the AS arm and three in the A arm). The mean relative dose intensities of the AS and A arms were 95.9% and 94.4%, respectively (p=0.667). The safety profiles of both treatment arms were similar (Table 3). There were no treatment-related mortalities in either arm. Common adverse events included skin rash (77.8% in the AS arm vs. 78.1% in the A arm), diarrhea (63.9% vs. 84.4%), and stomatitis (66.7% vs. 78.1%). Interestingly, more patients experienced grade 3 or 4 diarrhea in the A arm (5.6% vs. 18.8%). Close relationships between common adverse effects were observed. Patients experiencing grade ≥ 2 skin rash had a higher incidence of grade ≥ 2 stomatitis or diarrhea than those with grade < 2 skin rash (grade ≥ 2 stomatitis, 56.0% vs. 27.9%, p=0.022; grade ≥ 2 diarrhea, 48.0% vs. 27.9%, p=0.095). Additionally, there was a significant association between grade ≥ 2 stomatitis and grade ≥ 2 diarrhea (p=0.046).
4. EGFR status and clinical outcome
Among a total of 68 patients, the tumors of 64 patients were available for EGFR mutation analysis, 61 for the FISH test, and 56 for IHC staining analysis. EGFR mutations were detected in four of 64 cases (6.3%) (one exon 19 deletion, one exon 20 G810S, one exon 20 V786M, and one exon 21 G863S) (Table 4). Of the 61 tumors suitable for EGFR/FISH analysis, five (8.2%) were FISH-positive (two high polysomy, one gene amplification, and two both). Upon EGFR IHC analysis, positive expression was shown in 32 patients (57%) (16 for 2+ and 16 for 3+). There was no difference in the rate of EGFR mutation (5.6% vs. 6.2%), FISH-positivity (8.3% vs. 6.2%), or IHC-positivity (47.2% vs. 46.9%) between arms. In addition, the rate of these biomarkers did not differ with age, sex, ECOG PS, pathology (squamous cell carcinoma vs. others), or smoking status. There was no overlap between EGFR mutation and FISH positivity.
Upon analysis of all patients, EGFR mutation and EGFR/FISH status were significantly associated with the response to treatment, including afatinib, whereas EGFR IHC status was not. The EGFR-mutant patients showed higher ORR than the EGFR wild-type patients (50.0% vs. 5.0%, respectively; p=0.028) (Table 4). In addition, the EGFR/FISH-positive patients showed higher ORR than the EGFR/FISH-negative patients (40.0% vs. 5.4%, respectively; p=0.049). Thus, the EGFR-mutant or EGFR/FISH-positive patients (n=9) showed significantly higher ORR than those with tumors harboring both WT EGFR and EGFR/FISH-negativity (n=52) (44.4% vs. 1.9%, respectively; p=0.001). However, the EGFR mutation and EGFR/FISH status were not correlated with median PFS for treatment. The median PFS was 2.9 months (95% CI, 0.1 to 6.2) in the EGFR-mutant patients vs. 1.9 months (95% CI, 0.9 to 2.9) in the WT EGFR patients (HR, 0.42; 95% CI, 0.13 to 1.36; p=0.122), and 3.6 months (95% CI, 0.1 to 9.2) in the EGFR/FISH-positive patients versus 1.9 months (95% CI, 0.9 to 2.9) in the EGFR/FISH-negative patients (HR, 0.97; 95% CI, 0.38 to 2.45; p=0.944). The EGFR IHC status was not related to median PFS (IHC-positive vs. IHC-negative; 1.9 months vs. 3.6 months; p=0.976).
When the analysis was limited to 60 patients with tumors harboring WT EGFR, the ORR was 6.3% for the AS arm versus 3.6% for the A arm (p=1.000), median PFS was 1.0 versus 3.6 months (p=0.218), and median OS was 10.0 months versus 6.8 months (p=0.815). In the subgroup with EGFR-mutant tumors (n=4), the ORR of the AS versus A arm was 0.0% versus 100.0% (p=0.333), the median PFS was 1.9 months versus 2.9 months (p=0.433), and the median OS was 6.7 months versus 7.1 months (p=0.815). In terms of EGFR FISH status, the negative subgroup (n=56) had an ORR of 3.3% versus 7.7% (AS vs. A, p=0.592), a median PFS of 1.0 versus 3.6 months (p=0.209), and a median OS of 10.0 months versus 7.1 months (p=0.318), whereas the positive subgroup (n=5) had an ORR of 33.3% vs. 50.0% (AS vs. A, p=1.000), a median PFS of 4.0 vs. 0.9 months (p=0.207), and a median OS of 6.0 months versus 4.9 months (p=0.586). The subgroup analysis with EGFR-IHC negative tumors (n=4) had an ORR of 0.0% vs. 10.0% (AS vs. A, p=0.417), a median PFS of 1.0 months versus 3.6 months (p=0.789), and a median OS of 11.0 months versus 7.0 months (p=0.918), whereas the positive subgroup analysis had an ORR of 11.8% versus 13.3% (AS vs. A, p=1.000), a median PFS of 1.0 versus 1.9 months (p=0.122), and a median OS of 7.1 months versus 13.3 months (p=0.329).
5. Adverse effects as a predictive marker
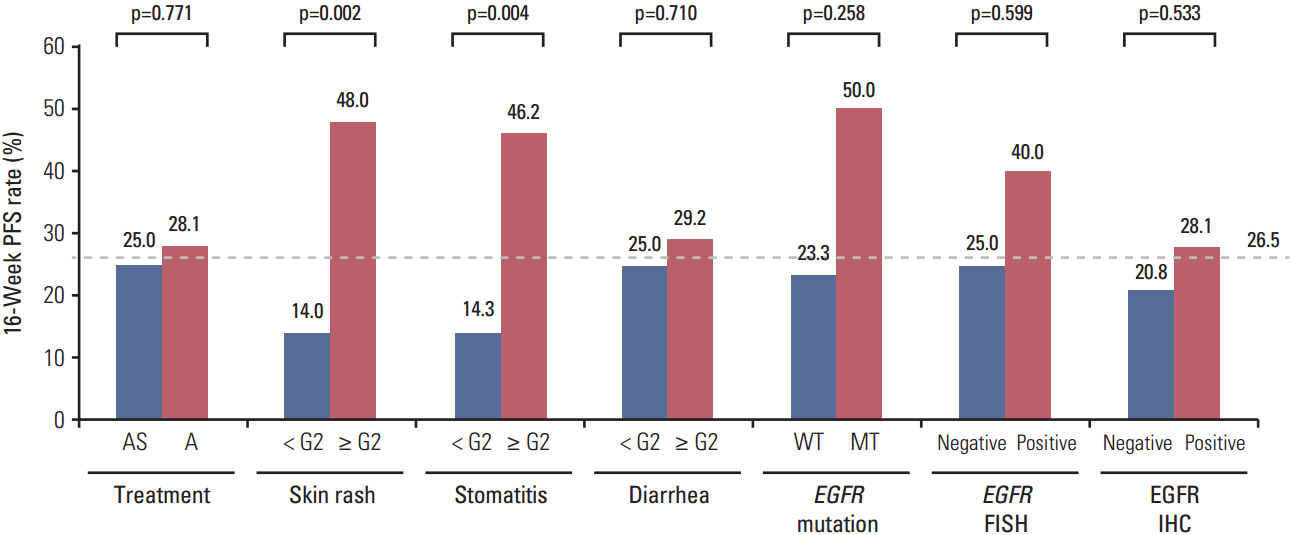
For all patients receiving afatinib, the adverse effects skin rash, stomatitis, and diarrhea were not significantly associated with the ORR, regardless of whether the patients received simvastatin. However, for the 16-week PFS rate, the grade of skin rash and stomatitis was significantly related to the efficacy of afatinib (Fig. 3). Patients who experienced grade ≥ 2 skin rash or stomatitis showed a significantly higher 16-week PFS rate than those with skin rash or stomatitis of grade < 2 (skin rash, 48.0% vs. 14.0%, p=0.002; stomatitis, 46.2% vs. 14.3%, p=0.004). These results remained unchanged in the landmark analysis at 4 weeks (skin rash, 57.1% vs. 20.0%, p=0.006; stomatitis, 54.5% vs. 20.7%, p=0.012). Upon multivariate analysis of PFS, only the skin rash grade was significantly related to the risk of progression (HR for skin rash grade ≥ 2 vs. grade < 2, 0.44; 95% CI, 0.25 to 0.78; p=0.005) (Table 5). When analysis was limited to rash-evaluable patients who continued treatment until at least 4 weeks, the difference in the progression of risk between groups remained unchanged (HR for skin rash grade ≥ 2 vs. grade < 2, 0.42; 95% CI, 0.22 to 0.81; p=0.010).
The dose reduction of afatinib was not significantly related to the ORR (yes vs. no, 14.3% vs. 5.6%, p=0.575). The risk of progression was the same in patients taking a reduced dose (HR for yes vs. no, 1.01; 95% CI, 0.54 to 1.89; p=0.967).
Discussion
To the best of our knowledge, this is the first clinical study to evaluate the emerging role of statins in overcoming resistance to EGFR-TKIs in patients with NA-NSCLC, which is usually resistant to EGFR-TKI therapy. The positive findings observed in the subgroup analysis of a previous clinical study were not confirmed in the present study [13]. The combination of AS in patients with NA-NSCLC did not improve the tumor response or survival outcomes compared with afatinib alone, although it did not increase the toxicities. These results were consistent with those observed for patients with tumors harboring WT EGFR.
Afatinib was recently approved for pretreated advanced NSCLC based on its superior efficacy compared with erlotinib in the LUX-Lung 8 trial [23]. The LUX-Lung 8 was a phase III randomized controlled trial that enrolled 795 pretreated patients with advanced squamous NSCLC globally to receive either erlotinib or afatinib, with a primary endpoint of PFS. The median PFS was significantly longer for afatinib than for erlotinib, (2.4 months vs. 1.9 months; HR, 0.82; p=0.0427), as was the median OS (7.9 months vs. 6.8 months; HR, 0.81; p=0.0077). Although this global trial was the most recent positive study of patients with squamous cell lung carcinoma, the clinical benefits of the superior arm were also modest. Thus, the role of predictive markers in selecting patients who are likely to benefit more from EGFR-TKIs is more important in this lung cancer population. Our study searched for predictive markers for afatinib treatment in the entire study population with NA-NSCLC. There were no biomarkers related to the EGFR pathway that were predictive of RR and PFS for afatinib treatment. The EGFR mutation and EGFR/FISH status were significantly associated with RR, but not median PFS, whereas EGFR IHC status was not related to either RR or PFS. Interestingly, the most common adverse event for afatinib, skin rash, was significantly associated with risk of progression in this study. This finding was consistent with the landmark analysis at 4 weeks, which was measured because skin rash generally develops after 1 week and reaches maximum severity after 2 to 3 weeks of EGFR-TKI treatment. Although several retrospective studies suggested that rash severity was associated with response or survival to EGFR-TKI treatment in NSCLC patients, a prospective study to evaluate the severity of skin rash as a surrogate marker for EGFR-TKI efficacy is needed [24-26]. Indeed, such a study will help guide EGFR-TKI treatment in NSCLC with WT EGFR, which lacks reliable predictive markers for EGFR-TKI efficacy.
The type, incidence, and severity of adverse effects in this study were comparable with those observed in another study with afatinib [23]. In the present study, grade ≥ 3 skin rash, stomatitis, and diarrhea developed in 2.9%, 2.9%, and 11.8% of patients, respectively, whereas they developed in 6.0%, 4.0%, and 10% of patients in a LUX-Lung 8 study [24]. In the present study, 26.6% of all patients reduced their afatinib dose, although the dose reduction did not affect the efficacy. Patients who reduced their afatinib dose to 30 mg/day did not show inferior clinical outcomes compared with those who did not. These findings suggest that the afatinib dose can be adjusted in patients experiencing potentially challenging side effects.
Interestingly, one patient showed a CR to afatinib for a long duration, even though he had squamous cell carcinoma NSCLC. The histological type was squamous cell carcinoma in both surgical specimens of the primary tumor and right adrenal gland metastasis. An EGFR exon 19 deletion mutation was detected in his resected lung tumor sample. Currently, the role of the EGFR mutation test in lung squamous cell carcinoma remains controversial; thus, the guidelines for this test are not consistent among oncology societies. Although activating mutations are rare in lung squamous cell carcinoma (< 5%), the treatment outcome with EGFR-TKI in squamous cell carcinoma with the EGFR mutation is not inferior to that in adenocarcinoma with the EGFR mutation, as shown in the CR case in our study [25,27]. Thus, for personalized medicine with comprehensive genomic analysis, an EGFR mutation test also should be included to select the correct drugs for patients with lung squamous cell carcinoma.
There are several issues to consider in interpreting the negative results of this study. First, the median PFS of the A arm in the present study was longer than that of the afatinib arm in the LUX-Lung 8 trial (3.6 months vs. 2.4 months). Five patients (15.6%) in the A arm withdrew from our study early; thus, the results may have been affected by this considerable number of censored events. In contrast, the median PFS of the AS arm in the present study was shorter than that of the afatinib arm in the LUX-8 trial (1.0 months vs. 2.4 months). This finding may be due to earlier assessment of the first tumor (4 weeks in this study vs. 8 weeks in the LUX-8 trial). Secondly, the dose of simvastatin used in this clinical study may have been too low to exert an anticancer effect when compared with the statin dose in preclinical studies. Despite the in vitro mechanistic rationale of statin studies, they have often been criticized for using high concentrations of statins (1-200 μM) because the maximum statin concentrations in the serum of patients receiving standard doses for hyperlipidemia therapy are 10-200 nM [28,29]. Whether the statins at higher doses confer therapeutic benefits to cancer patients has yet to be determined. Additionally, the resistance mechanism for EGFR-TKI of NA-NSCLC may be more complicated than RAS activation and downstream signaling cascades, which are expected to be inhibited by statins. In another phase II study of patients with KRAS mutant refractory colorectal tumors, adding simvastatin to cetuximab and irinotecan produced highly favorable clinical outcomes with a disease control rate of 65.4%, a PFS of 7.6 months, and an OS of 12.8 months [30]. Thus, the combination of statins and EGFR-blocking agents may be effective in specific tumor types that are dependent on RAS and its downstream pathways.
The addition of simvastatin to afatinib did not improve efficacy versus afatinib alone in pretreated patients with NA-NSCLC. However, no unexpected adverse events were observed in response to treatment with combined AS. EGFR mutation, EGFR FISH, and EGFR IHS status were not reliable as predictive markers for afatinib efficacy. However, skin rash severity may be useful for making treatment decisions regarding afatinib in this population.