Introduction
Despite increasing evidence showing that targeted therapy against the molecular drivers of cancer has the potential to improve the survival of cancer patients [1], standard therapy with a “one size fits all” strategy involving cytoreductive surgery and platinum/taxane-based chemotherapy is still being used in patients with advanced epithelial ovarian cancer (EOC) without consideration of molecular phenotype or biomarkers [2]. Accordingly, new therapeutic strategies for precision medicine are urgently needed in EOC in particular because of the severity of the disease and its clinical heterogeneity.
Currently, experimental cancer therapy has primarily involved evaluation of drugs using human cancer cell lines in vitro and their xenograft models. However, because established cancer cell lines have irreversibly lost important biological properties of their original organ site, the cell lines and their xenograft models do not exactly reflect the original phenotypic or genomic characteristics of various cancer types [3,4]. In contrast to cell lines and their xenografts, patient-derived xenografts (PDXs) can be established directly from a patient’s tumor tissue without prior in vitro culture by immediately transplanting tumor tissue excised at the time of surgery into immunodeficient mice. Because they can accurately recapitulate the complexity and heterogeneity of the patient’s tumor, PDX models can be considered powerful tools for preclinical studies of targeted therapeutic strategies that narrow the gap between laboratory bench discoveries and clinical translation [2]. The value of PDX is emphasized in precision medicine for cancer types with a heterogeneous nature. PDX models have been applied to preclinical drug testing and biomarker identification in several cancers, including breast, lung, pancreatic, brain, and colon cancers [5].
Recent studies of EOC have reported various conditions and approaches for the development of PDX models, either orthotopically using intraperitoneal (IP) and intra-ovarian bursa methods, or nonorthotopically using subrenal capsule, subcutaneous (SQ), and mammary fat pad (MFP) methods. These studies have resulted in varied success rates and genomic or phenotypic similarities [2]. Two previous studies suggested that PDXs might be successfully developed via a subrenal implantation method [6,7], although they did not perform molecular testing. The present study was designed to develop a PDX model for EOC with pathological and molecular similarities to the primary patient tissue using a subrenal implantation method and apply this model to preclinical drug testing.
Materials and Methods
1. Patients and tissue specimens
Tumor samples from patients with EOC were taken from fresh surgical specimens immediately after primary or secondary debulking surgery at the Department of Obstetrics and Gynecology, Samsung Medical Center, Seoul, Korea between May 2011 and March 2014. This study was conducted with the Samsung Medical Center Institutional Review Board approval (IRB File No. 2009-09002) and carried out in accordance with the approved guidelines and regulations. All patients provided written informed consent authorizing the collection and use of tissues for study purposes. Clinical information was obtained from medical records, including age, stage, cell type, grade, optimality, response to chemotherapy, recurrence, and survival. Tumors were classified as EOC according to World Health Organization criteria based on examination by gynecologic pathologists. Patients with histologically confirmed invasive epithelial ovarian, peritoneal, or fallopian tube cancer were eligible for inclusion. Patients with borderline ovarian cancer, mesenchymal tumor, sex-cord stromal tumor, germ cell tumor, and Krukenberg tumor were ineligible for this study. Overall survival (OS) was defined as the time between histologic diagnosis and death or last follow-up. Progressionfree survival (PFS) was defined as the time between histologic diagnosis and first progression or recurrence, death as a result of disease, or last follow-up. Platinum resistance was defined as less than 6 months of platinum-free interval. Date of first progression was determined based on cancer antigen 125 levels and imaging results according to the Response Evaluation Criteria in Solid Tumors guideline ver. 1.1 [8]. Surgical outcome was categorized as no residual disease, optimal (< 1 cm), or suboptimal (≥ 1 cm).
2. Establishment of PDX model using subrenal implantation
Fresh tissues from consenting patients with ovarian cancer were collected at the time of debulking surgery at Samsung Medical Center, Seoul. Frozen sections were first analyzed by a pathologist to make the diagnosis of cancer. More than 3 cm3 of non-necrotic beside cancer tissue was obtained by clinical staff in the gynecology oncology department. Female BALB/c nude mice were purchased from Orient Bio (Seongnam, Korea). Within 2 hours of removal of tissues in the operating room, patient tumor specimens were reduced into small pieces (less than 2-3 mm), implanted into the subrenal capsule of the left kidney of mice (n=5 per tumor sample), and propagated by serial transplantation [6] (S1 video clip). All procedures, including tumor implantation and measurement of tumor size, were performed by one researcher (Y.J.C.). The mice used in these experiments were 6-8 weeks old and were monitored daily for tumor development. When the tumors reached approximately 2 cm at the grafted site or the mice became moribund, the animals were euthanized and the tumors were collected. These samples were considered tumorigenic and designated “M1.” Tumorigenic samples were serially passaged in vivo to generate “M2” PDX tumors. We recorded body weight, tumor weight, and number of tumor nodules. Tumors from PDXs were fixed in formalin and embedded in paraffin or snap-frozen in OCT compound (Sakura Finetek Japan, Tokyo, Japan) in liquid nitrogen. This study was approved by the Institutional Animal Care and Use Committee of the Samsung Biomedical Research Institute, which is an accredited facility of the Association for Assessment and Accreditation of Laboratory Animal Care International (protocol No. H-A9-003) and carried out in accordance with the guidelines approved in the Institute of Laboratory Animal Resources Guide. The success of PDXs was defined as the development of tumor in mice within 12 months of initial implantation.
3. Histologic examination
To compare histopathologic features of tumor tissues, hematoxylin and eosin (H&E) staining was performed on all paraffin blocks from tissue samples obtained from the primary patient tumor and PDX tumor. All microscopic observations and comparisons were performed by two gynecologic pathologists (S.Y.S. and Y.-L.C.).
4. Short tandem repeat
Short tandem repeat (STR) analysis was performed at 16 loci on different chromosomes as previously described [9] to verify that the PDX samples analyzed were derived from each patient. Target DNA (10 ng) was amplified by multiplex polymerase chain reaction (PCR) using fluorescent dyelinked primers for the 16 loci: 13 autosomal STR loci (CSF1PO, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11, FGA, TH01, TPOX, and vWA); two additional STR loci, D2S1338 and D19S433, and the amelogenin locus. Amplification was performed using an AmpFlSTR Identifiler PCR Amplification Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. PCR products generated were mixed with an internal size standard (GS-500 LIZ, Applied Biosystems), electrophoresed on an ABI 3130xL Genetic Analyzer (Applied Biosystems), and analyzed with the GeneMapper 4.0 software using the supplied allelic ladders (Applied Biosystems).
5. Western blot analysis
Immunoblotting was performed using a 1:1,000 dilution of anti–epidermal growth factor receptor (EGFR) antibody (#2646, Cell Signaling Technology, Beverly, MA) or a 1:3,000 dilution of anti–β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and horseradish peroxidase–conjugated anti-rabbit or anti-mouse secondary antibody. Bands were visualized with an enhanced chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer’s protocols.
6. Combination chemotherapy with paclitaxel and carboplatin in a PDX model
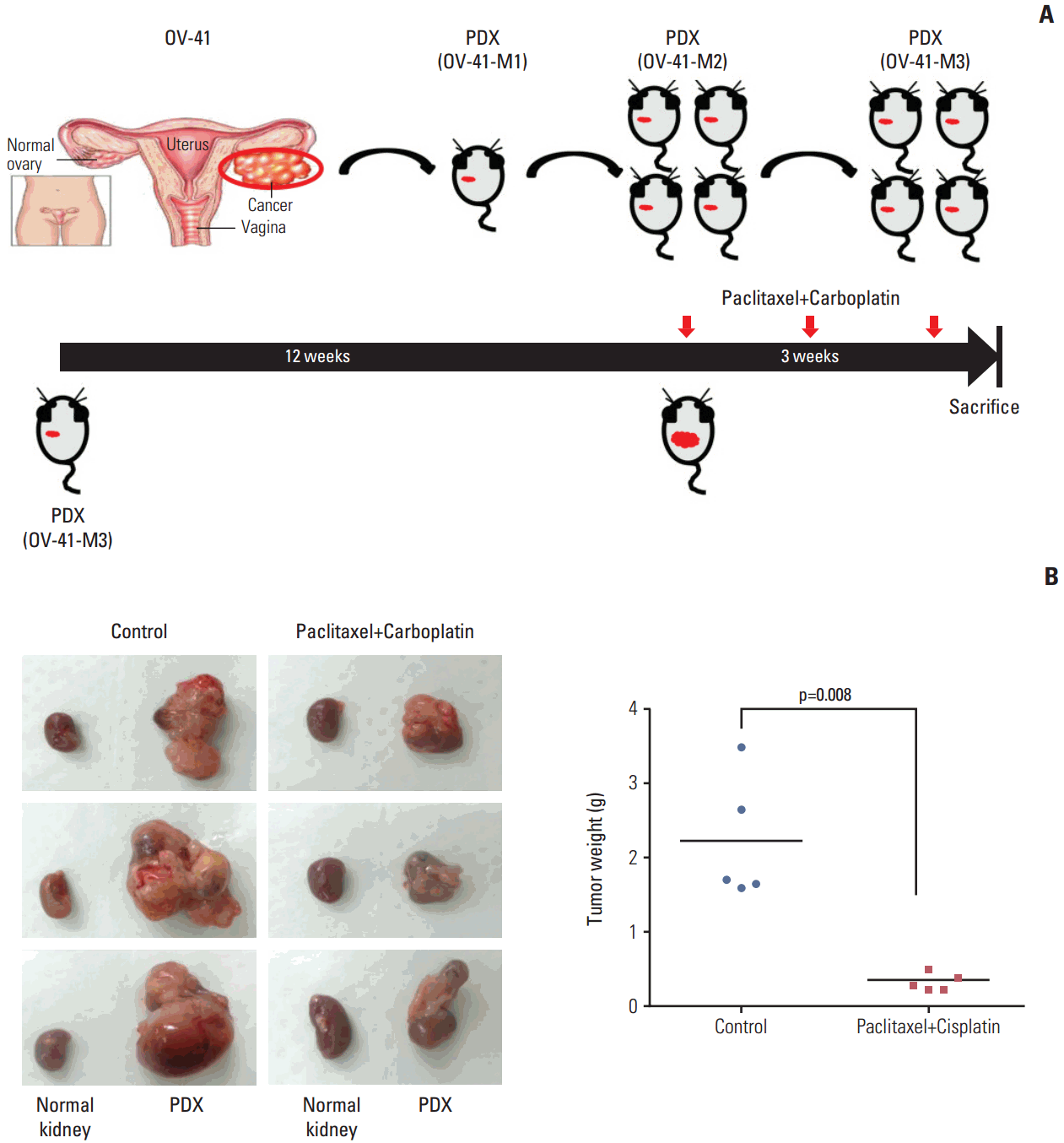
The PDXs (OV-41-M3) were used to test for response to combination chemotherapy with paclitaxel and carboplatin as used in clinical practice as a standard adjuvant regimen. The patient corresponding to the OV-41 sample was a 70-year-old woman who had stage IIIC, high-grade serous EOC. We treated the PDXs (OV-41-M3) for 3 weeks (on days 1, 8, and 18) starting 3 months after the implantation of xenograft tissues. Paclitaxel (15 mg/kg, BMS, Princeton, NJ) and carboplatin (50 mg/kg, Hospira, Chicago, IL) were administered intraperitoneally to the mice. Paclitaxel was administered 30 minutes prior to carboplatin. All drugs were administered as 0.9% saline solutions in a volume of 200 μL [10]. The mice used in these experiments were 6-8 weeks old. The mice (n=5 per group) were monitored daily for tumor development and response to treatment and any that appeared moribund were sacrificed.
7. EGFR inhibitor therapy in clear cell carcinoma cell line and PDX models
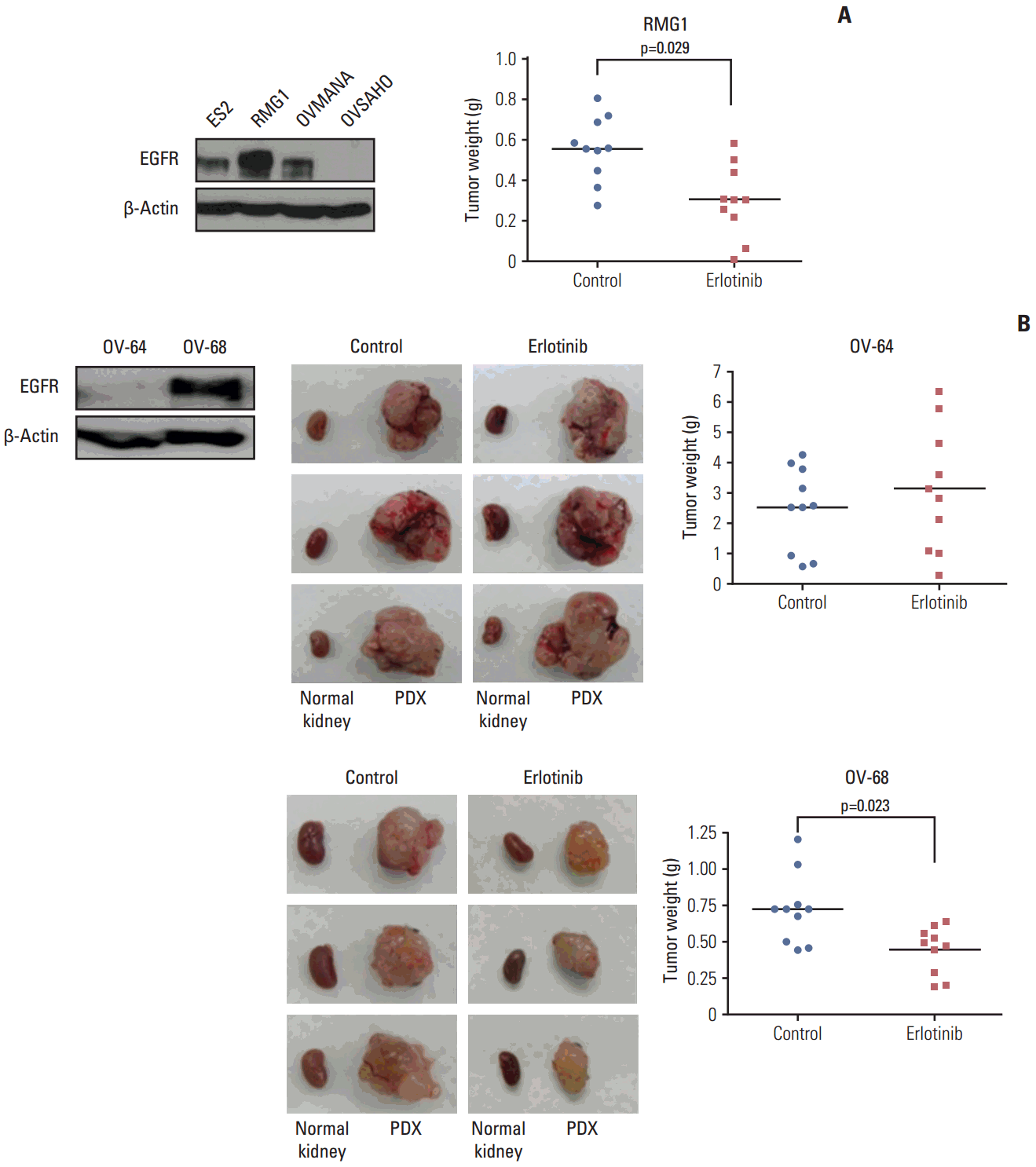
RMG1 cells were purchased from the JCRB cell bank (JCRB, Osaka, Japan) and maintained in complete media (Ham’s F12) supplemented with 10% fetal bovine serum and 0.1% gentamicin sulfate (Gemini Bioproducts, Calabasas, CA) in 5% CO2 at 37°C. Among the PDXs, we selected two models, OV-64 and OV-68, from patients with clear cell carcinoma histology. The mice were treated with erlotinib (25 mg/kg, Selleckchem, Boston, MA) by IP injection three times a week as previously described [11] starting 1 month after RMG1 injection or 4 months after the implantation of xenograft tissues. The mice used in these experiments were 6-8 weeks old. Mice (n=10 per group) were monitored daily for tumor development and response to treatment and were sacrificed when any appeared moribund. We recorded body weight, tumor weight, and number of tumor nodules. Tumors were fixed in formalin and embedded in paraffin or snap-frozen in OCT compound (Sakura Finetek Japan) in liquid.
8. Statistical analysis
The chi-squared or Fisher exact test was performed for comparisons between engraftment status and patient characteristics. OS and PFS according to engraftment status were assessed using Kaplan-Meier curves and the log-rank test. To adjust for clinical significantly prognostic variables, the Cox proportional hazards model was used for multivariate analysis of PFS. OS was not analyzed using the Cox proportional hazards model because of the extremely small number of death events (three patients). The SPSS software package ver. 18.0 (SPSS Inc., Chicago, IL) was used for all statistical analyses. A p-value of ≤ 0.05 was considered to indicate significance, and all p-values were two-sided.
Results
1. Establishment of EOC PDXs and their clinicopathological characteristics
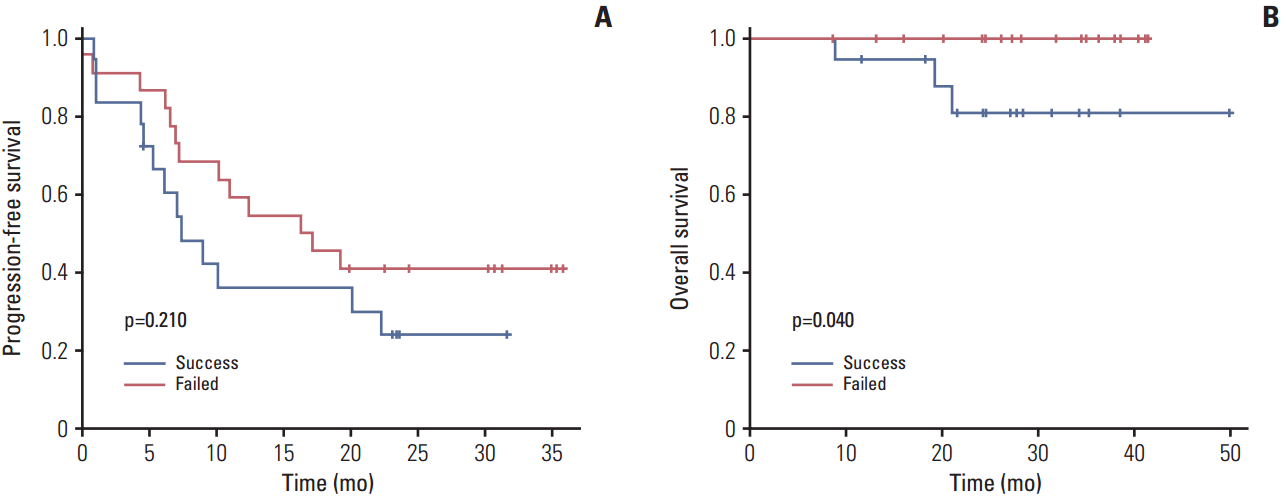
Surgical specimens were collected from 45 patients with EOC. in vivo tumorigenicity was defined as the development of tumor in mice within 12 months of initial implantation. Of the 45 patient samples implanted, 22 were successfully engrafted in mice to create PDX models (engraftment rate, 48.8%). The median duration of first generation of PDX development (M1) from initial implantation of patient tissues was 5.3 months (range, 1.9 to 12.0 months). Baseline clinicopathologic characteristics of the patients according to generation of PDX models are shown in Table 1. We analyzed the association between PDX formation and the clinical characteristics of the corresponding EOC patients. Patients whose tumors successfully engrafted in mice had significantly inferior OS (p=0.040) and a tendency toward inferior PFS (p=0.210) (Fig. 1). The platinum-resistant group had a strong association with successful engraftment compared with the sensitive group (p=0.023; hazard ratio [HR], 5.55; 95% confidence interval [CI], 1.27 to 24.29). However, with the exception of platinum response, there was no significant correlation between the in vivo tumorigenic potential and other clinical factors, including age, histological type, tumor grade, stage, timing of surgery, and optimality of primary debulking surgery (Table 1).
2. Histopathologic and genetic stability
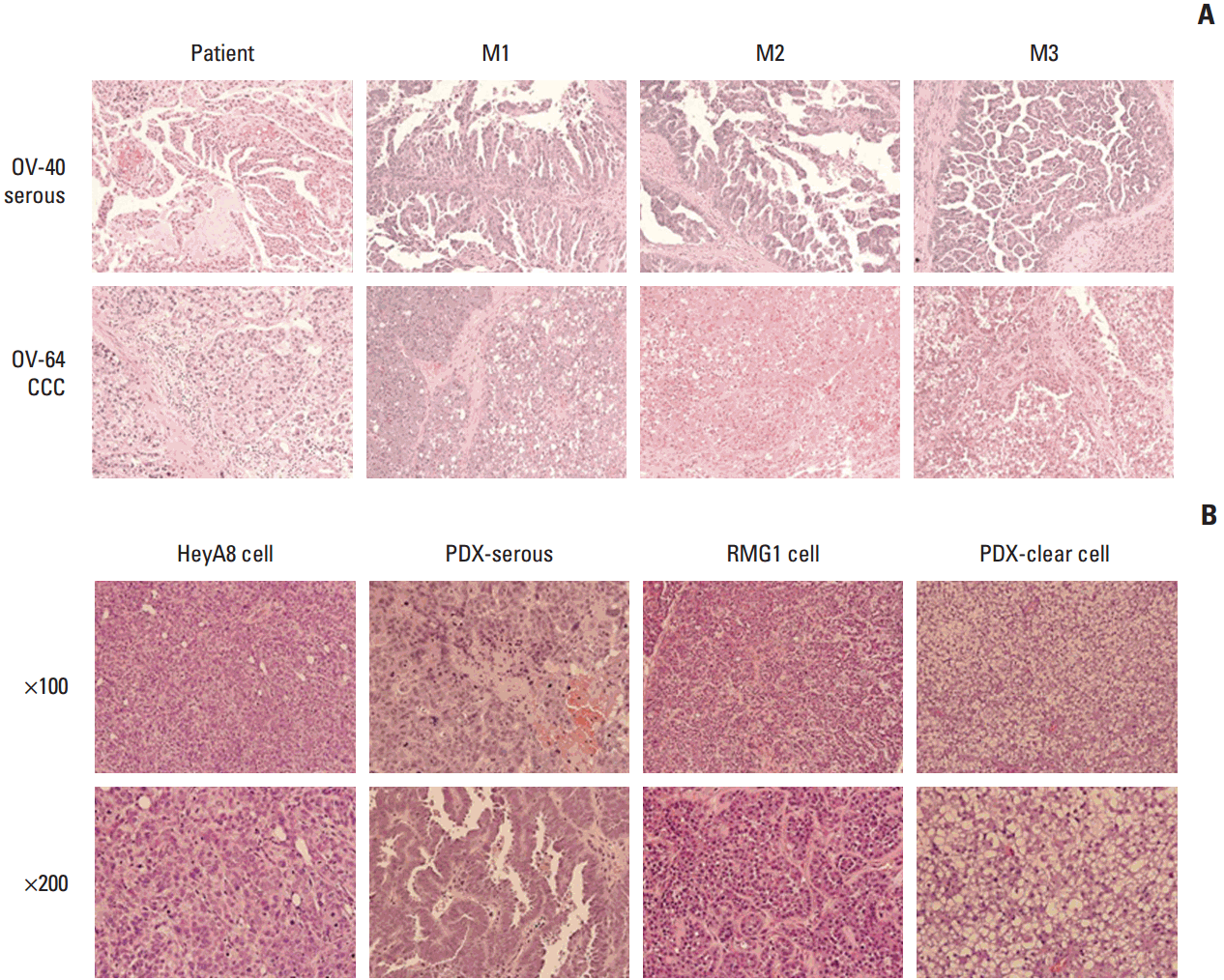
A comparison of the H&E staining of primary patient tissue and PDXs after each passage revealed a similar architectural pattern of nesting configuration and comparable cytologic atypia in all PDXs according to pathologic subtype (Fig. 2A). However, we found quite different histologic features in PDX tissues compared with tissues of cell line xenografts. Specifically, PDX tissues for serous histology showed papillary projection and stromal tissues, while those for clear cell histology had clear cytoplasm similar to the patient tissues, whereas these features were not seen in the corresponding cell line xenografts (HeyA8 and RMG1 respectively) (Fig. 2B).
We conducted quality control testing of the genomic status of the PDXs and the original patient tissues. STR analysis was performed at 15 loci on different chromosomes to verify that the PDXs were derived from each patient’s primary tissue (S2 Table). The banding patterns for primary patient tissue and PDXs were almost identical.
3. Representative example of a PDX model
Fig. 3 shows a representative case of EOC with high-grade serous type, which is the most common and refractory pathologic subtype. The duration of development of PDX from implantation to the next passage was relatively consistent, ranging from 5 to 6 months. H&E staining showed that the morphology and tissue architecture were preserved (Fig. 3A). STR (Fig. 3B) conducted for validation of the PDX model indicated that the original patient tumor and the PDX tumor had identical genetic characteristics. These data suggest that EOC PDXs derived using this approach can represent the original tumors.
4. Validation of the effect of paclitaxel-carboplatin combination chemotherapy in a PDX model
Combination chemotherapy with paclitaxel and carboplatin is still the standard regimen for EOC treatment, with an 80% response rate in the primary adjuvant setting. Therefore, we performed experiments to test for the response to combination therapy in the EOC PDXs model for high-grade serous ovarian carcinoma (OV-41). Paclitaxel-carboplatin chemotherapy significantly decreased the tumor weight in PDXs (OV-41-M3) compared with the phosphate buffered saline control (p=0.008) (Fig. 4). The patient corresponding to the OV-41 PDXs model had undertaken primary debulking surgery, after which he achieved no residual cancer.
The patient received six cycles of adjuvant combination chemotherapy with paclitaxel and carboplatin, showed complete remission upon computed tomography scan after three cycles of combination chemotherapy, and was alive with no evidence of disease after a follow-up period of 18 months. Therefore, the patient-specific response to standard treatment could be reproduced in the PDXs model.
5. Therapy experiments of EGFR inhibitor in the PDXs of clear cell carcinomas
In a recent report, among different EOC histologies, clear cell carcinoma showed activation of EGFR as a distinct signaling network compared to serous or endometrioid types [12]. Therefore, we tested the efficacy of the EFGR inhibitor in the cell line and PDX model for clear cell carcinoma. Measurement of baseline expression of EGFR protein using western blotting in EOC cell lines (ES2, RMG1, OVMANA, and OVSAHO) and PDXs tissues (OV-64 and OV-68) of the clear cell type showed that ES2, RMG1, OVMANA, and PDXs (OV-68) cells expressed EGFR protein. In an orthotopic cell line xenograft model of RMG1, which had the strongest expression of EGFR, treatment with the EGFR inhibitor erlotinib significantly decreased tumor weight compared with the control (p=0.029) (Fig. 5A). Moreover, erlotinib significantly decreased tumor weight in the OV-68 PDXs model, which also strongly expressed EGFR compared with the control (p=0.023). In contrast, the OV-64 PDXs model with relatively low expression of EGFR did not show the erlotinib effect (Fig. 5B).
Discussion
Different sites of implantation in the mouse including SQ, IP, the orthotopic intra-ovarian bursa, and the subrenal capsule have been tested to establish PDXs of EOCs. Both orthotopic and heterotopic ovarian PDX models have reported similar histology, genetic and molecular expression profiles, and overall tumor phenotype to that of the primary patient tumor [13]. For EOCs, engraftment rates has been reported to be 25%-95% depending on the cell type and method [6,14-17] and growth usually takes 4-8 months [18]. In the present study, we obtained an engraftment rate of 48.6% (22/45 cases) using the subrenal implantation method as previously described [6] in female BALB/c nude mice and the median time to development of the first generation in mice was 5.3 months (range, 1.9 to 12.0 months).
According to recent reports for the development of PDX, EOCs reportedly have high engraftment rates of 74% [17] to 85% [16] or low rates of 25% [15] to 40% [19]. There are several possible factors responsible for this wide range of successful engraftment, including (1) quality of patient tumor tissues, including size of tissues, tumor percent in samples, and the portion of necrosis in tumors; (2) location of implantation site (SQ, IP, orthotopic intra-ovarian bursa, or subrenal capsule); and (3) type of immuno-deficient mice (nude or NOD/SCID). Moreover, the lag of physical time from collecting tissues during surgery to implanting in mice is another important factor influencing successful implantation. The location of implantation and type of immuno-deficient mice are also important to the PDX engrafting rate. Dobbin et al. [13] compared the rate based on the location of implantation as SQ, MFP, IP, and subrenal capsule in SCID mice. They reported a PDX engraftment rate of 85.3% SQ, 63.64% MFP, 22.2% IP, and 8.3% in the subrenal capsule [13]. However, Lee et al. [6] reported an engraftment rate of more than 95% following subrenal capsule implantation into SCID mice. Moreover, Topp et al. [16] reported an 83% engraftment rate by SQ and intrabursa fat pad implantation into SCID mice, while Weroha et al. [17] reported a 75% engraftment rate by IP implantation into SCID mice, Zhu et al. [19] reported a 40% engraftment rate by ovarian capsule implantation into nude mice and Ricci et al. [15] reported a 25% engraftment rate by SQ and IP implantation into nude mice. We initially attempted to implant tumor tissues to various routes including SQ, IP, the intrabursa fat pad, and the subrenal area in a pilot study (data not shown) and found that subrenal implantation was the most successful and easiest method for this model.
The type of immuno-deficient mice can also affect the engraftment rate of PDX. A better engraftment rate could probably be achieved by implanting tumor tissues into SICD or NOD-SCID-IL2γR mice, rather than BALB/c nude mice [16,17]. In the present study, we used BALB/c nude mice for the PDX model because these are less expensive than SICD or NOD-SCID-IL2γR and we did not find large differences in the success rate between the two types of mice through our pilot test.
Preclinical models to predict those patients who will benefit from targeted therapy are imperative to implementation of effective precision therapy strategies. Although the utility of PDX models is still being evaluated, it has been reported that PDX tumors show preservation of cytogenetics, cellular complexity, and glandular, vascular, and stromal architecture compared to the primary patient tumor [20,21]. Furthermore, genetic stability between PDXs and the primary patient tumor was demonstrated [7,13,17]. In the present study, a quality control process using H&E staining and STR testing showed highly similar histopathologic features and molecular features. As a result, we demonstrated that the developed PDXs accurately represented the histopathologic and genetic phenotype of the primary patient tumor. However, in this study, we did not demonstrate genomic analysis between donor patients and corresponding PDX tissues to identify genetic similarity. This is a major limitation to our model because most published PDX studies have reported cancer genome analysis between patient and PDX tumors relevant to biology or clinical response to therapy. Accordingly, additional detailed genetic analysis of this model should be conducted.
Annotation of clinical data to preclinical data could be helpful to interpretation of the results of studies conducted using PDXs models to predict the clinical course and response to novel target agents. Weroha et al. [17] previously reported annotation of patient clinical data to PDXs of ovarian cancer. In the present study, we found that patients whose tumors successfully engrafted in mice had inferior OS (p=0.040) (Fig. 1). Thus, these results implied that in vivo PDXs tumorigenicity was associated with inferior survival outcome of the corresponding patient, consistent with the results of previous studies [17]. Moreover, platinum resistance of EOCs had a strong association with successful engraftment (p=0.023; HR, 5.55; 95% CI, 1.27 to 24.29). Considering that the demand for PDXs for preclinical testing is greatest in the case of platinum-resistant EOC, this association between tumorigenicity and platinum resistance reinforces the utility of PDXs as a platform for preclinical testing of novel therapies for EOC. The response to therapy is important to validate the clinical predictive value of PDX models. The response to paclitaxel-carboplatin chemotherapy in PDXs was concordant with the clinical response of the corresponding patient (Fig. 4). Moreover, erlotinib significantly decreased the tumor weight in PDXs of clear cell carcinoma with overexpression of EGFR (Fig. 5). However, we only conducted drug tests for PDXs of selected cases because of cost and time constraints. While this is a limitation of the study, the purpose of our study was to establish PDX models of EOC and demonstrate their potential as a platform for translation research. Although, we only tested a few cases using this PDX platform, our results are important because they suggest that use of PDX models should be widened. The effects of erlotinib in PDXs of clear cell carcinoma with overexpression of EGFR should be interpreted only as an preclinical data. To date, no clinical studies have confirmed the effectiveness of erlotinib in EOC [22]. The present study was also limited in that erlotinib was tested in only two PDXs models of clear cell carcinoma, preventing generalization of the results. Nevertheless, the findings still demonstrate that PDXs have potential for analysis of tumor development and adaptation, screening of novel targeted therapies, testing of drug efficacy, identification of biomarkers, predicting clinical outcomes, and development of individualized treatment plans. Future studies to further validate and test this PDXs library are required.
The PDXs models themselves also have some limitations that may hamper their utility, primarily high cost, requirement for specialized skills, and the long latency to generate PDXs [18]. Although the PDXs model exists as a platform for conducting precision medicine, most patients do not have time to wait for a model of their tumor to undergo drug response testing. Improving engraftment rates, decreasing the latency between engraftment time and initiation of patient treatment, and new engraftment techniques using smaller tissue samples are required to widen the use of PDXs models. Moreover, because immunocompromised mice are used to generate PDXs, these models do not provide a platform to understand the role of the immune system when testing vaccines, immunomodulators, and immune activating therapies [18]. Despite these challenges to the use of the PDXs model, constant efforts to overcome the hurdles and develop personalized treatments for gynecologic malignancies using these models are ongoing.
In conclusion, PDXs models for EOC that have histopathologic and genetic stability can be efficiently developed by subrenal capsule implantation. These models have the potential to provide a promising platform for translational research and precision medicine in EOC.