Introduction
BRAF somatic mutations that render BRAF constitutively active are observed in 50%-60% of malignant melanomas [1]. Thus, BRAF inhibitors have recently shown promise for the treatment of metastatic melanoma harboring such BRAF mutations [2]. We also reported UAI-201 (also known as UI-152) as a potent ATP-competitive inhibitor of RAF proteins [3]. UAI-201 is more than 1,000-fold more selective at inhibiting the proliferation of tumor cell lines bearing the BRAF V600E mutation when compared with that of cells carrying wild-type BRAF [3]. However, the development of acquired resistance to inhibitors of oncogenic BRAF limits the duration of the tumor response [4]. Besides BRAF inhibitors, most anticancer drugs have the problem of drug resistance, which limits their effectiveness. Accordingly, understanding the molecular mechanisms of drug resistance is necessary to improve the effectiveness of cancer therapies. In general, reactivation of the mitogen-activated protein kinase (MAPK) pathway is considered a primary mechanism underlying the acquired resistance to BRAF inhibitors [5]. Our previous study indicated that induction of resistance to a BRAF inhibitor is associated with the inability of Spry2 to inhibit BRAF V600E activity in cells with mutant BRAF [6]. In fact, the relief of feedback after targeted therapy may be viewed as a key contributor to therapeutic resistance [7].
Small noncoding microRNAs (miRNAs) have been confirmed to regulate the expression of target mRNAs by repressing their translation [8]. A growing body of evidence shows that dysregulation of miRNA expression contributes to acquisition of drug resistance by cancer cells [9]. Nevertheless, relatively few studies have explored the roles of miRNAs in resistance to BRAF inhibitor therapy, although several studies identified miRNAs that alter some of the oncogenic factors in melanoma cells [10]. In particular, overexpression of miR-514a inhibits NF1 expression, which is correlated with increased survival of BRAF V600E cells treated with PLX4032 [11].
In this study, we used the Affymetrix miRNA V3.0 microarray profiling platform to compare miRNA expression levels in three melanoma cell lines: BRAF inhibitor–sensitive A375P BRAF V600E cells, their BRAF inhibitor–resistant counterparts (A375P/Mdr), and SK-MEL-2 BRAF wild type (WT) cells. The A375P/Mdr cells with acquired resistance to BRAF inhibitors were generated by propagating parental A375P cells harboring the BRAF V600E mutation at increasing concentrations of a BRAF inhibitor to implement chronic selection [12]. The SK-MEL-2 cell line expressing WT BRAF has intrinsic resistance to BRAF inhibition because the BRAF inhibitor lacks activity against cell lines that express WT BRAF. We found that 43 miRNAs are deregulated in BRAF inhibitor–resistant cells compared with those in BRAF inhibitor–sensitive cells. We also found that ectopically expressed miR-1246 can confer resistance to PLX4720 (a BRAF inhibitor) to A375P/Mdr cells.
Materials and Methods
1. Materials
The Affymetrix miRNA V3.0 array profiling platform was supplied by Affymetrix (Santa Clara, CA). The RNeasy Midi Kit was acquired from Qiagen (Valencia, CA). SYBR Premix EX TaqII, which was used for real time polymerase chain reaction (PCR), was purchased from Takara Korea Biomedical Inc. (Seoul, Korea). For the apoptosis assay, a FITC Annexin V Apoptosis Detection Kit was purchased from BD Biosciences Pharmingen (San Diego, CA). For the flow cytometric autophagy assay, Cyto-ID Green dye was acquired from ENZO Life Sciences, Inc. (Farmingdale, NY). Rabbit polyclonal anti-MEK, anti-ERK, anti-p21Cip1, and anti-p27Kip1 antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA), whereas anti–phospho-MEK (anti–p-MEK Ser217/221) and anti–phospho-ERK (anti–p-ERK, Thr202/Tyr204) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and penicillin-streptomycin were purchased from Life Technologies (Carlsbad, CA). The reagents for sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis were acquired from BioRad (Hercules, CA), while PLX4720 was obtained from Selleck Chemicals (Houston, TX).
2. Cell lines and cell culture
For this study, we used three melanoma cell lines: BRAF inhibitor–sensitive A375P BRAF V600E cells, their BRAF inhibitor–resistant counterparts (A375P/Mdr), and SK-MEL-2 BRAF-WT cells with intrinsic resistance to the BRAF inhibitor. A375P and SK-MEL-2 cells were acquired from either the Korean Cell Line Bank (KCLB; Seoul, Korea) or YOUAI Co., Ltd. (Suwon, Korea). A375P/Mdr cells were previously established by chronic selection with increasing doses of an inhibitor of oncogenic BRAF [12]. All cell lines were maintained at 37°C in DMEM supplemented with 10% FBS, penicillin-streptomycin, and glutamine. A375P/Mdr cells were further propagated in growth medium containing the BRAF inhibitor PLX4720 (1 μM). Before their use in the experiments, A375P/Mdr cells were maintained in PLX4720-free culture medium and subcultured at least three times. For experimental purposes, the cells were cultured in 60-mm tissue culture dishes until they reached ~80% confluence. PLX4720 was dissolved in dimethyl sulfoxide (DMSO) and diluted immediately before each experiment. The DMSO concentration was less than 0.1% in all experiments.
3. MiRNA microarray profiling
Total RNA was extracted from three melanoma cell lines using a miRNeasy Kit (Qiagen). The quantity and purity of RNA samples were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Total RNA was subjected to miRNA expression profiling (miRBase release 17.0) using the Affymetrix miRNA V3.0 array with a probe set for 1,733 human mature miRNAs. Microarray hybridization, data generation, and normalization were performed by GenoCheck Co., Ltd. (Seoul, Korea). The Affymetrix Expression Console software and Affymetrix 7G scanner were used for bioinformatic analysis and visualization of microarray data.
4. Real-time quantitative reverse-transcription PCR
Real-time quantitative reverse-transcription PCR (qRTPCR) was performed to confirm the expression of differentially expressed miRNAs that were identified in the miRNA microarray analysis. Briefly, total RNA was isolated using a RNeasy Mini Kit (Qiagen). The primer sequences were then selected using the Primer3Plus software (http://primer3plus.com/), after which the primers were synthesized by Bioneer (Daejeon, Korea). The primers used in the present study had the following oligonucleotide sequences: miR-3617, 5′-CTCAAAGGGCTCATTGGTTG-3′ and 5′-GCCTGCTAAATGGAGTGTGC-3′; miR-92a-1, 5′-GCCCAATCAAACTGTCCTGT-3′ and 5′-CAATCCCCACCAAACTCAAC-3′; miR-1246, 5′-TGAAGTAGGACTGGGCAGAGA-3′ and 5′-TGTTTGCAATAGCCCTTTGAG-3′; miR-193b-3p, 5′-ATCCCGGTTCTCCAAACTCT-3′ and 5′-TGGTAGCTCTCTGCCCTCAC-3′; miR-17-3p, 5′-TTGTAAAACTGAAGATTGTGACCA-3′ and 5′-TGCCAGAAGGAGCACTTAGG-3′. qRT-PCR was conducted on an Applied Biosystems 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA) using SYBR Premix EX TaqII. The mean threshold cycle (Ct) was calculated from data from triplicate reactions. The 2−ΔΔCt method was used to calculate the fold differences in miRNA among the tested samples [13].
5. miRNA mimic transfection procedures
The sequence of the miR-1246 mimic (5′-AAUGGAUUUUUGGAGCAGG-3′) was retrieved from the miRBase database (http://www.mirbase.org), and the sequence of the miR-122 inhibitor was completely complementary to that of miR-1246. Cells were transiently transfected with 50 nM miR- 1246 mimic, inhibitor or its negative control (Bioneer) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
6. Cell growth assay
Cells were transfected with either miRNA mimics or inhibitors and their control using Lipofectamine 2000 for 24 hours. The cells were plated in quadruplicate in 96-well microtiter plates (Costar, Cambridge, MA) at 5×103/well and then incubated with PLX4720 at 37°C in a humidified 5% CO2 incubator. On day 3, the cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at 37°C for 3 hours. The absorbance of the samples against a background control (medium alone), which served as a blank, was measured at 450 nm using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA).
7. Flow cytometric analysis of autophagic activity with Cyto-ID staining
The Cyto-ID fluorescent reagent specifically labels autophagic vacuoles and colocalizes with LC3 [14]. Thus, autophagosome formation in cells was detected using the Cyto-ID Autophagy Detection Kit (ENZO Life Sciences Inc.). Briefly, cells were trypsinized, pelleted by centrifugation, and washed twice in a phenol-red-free culture medium with 2% FBS. The cells were then resuspended in 0.5 mL of freshly diluted Cyto-ID reagent and incubated at 37°C for 30 minutes, after which the Cyto-ID fluorescence of cells was immediately analyzed using a Gallios flow cytometer and the Kaluza analysis software (Beckman Coulter Inc., Brea, CA). The percentage of cells with Cyto-ID staining was used to assess the formation of autophagosomes.
8. Apoptosis assay using annexin V staining and flow cytometry
Apoptosis was quantified using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences Pharmingen). Briefly, after treatment with the BRAF inhibitor, 2×106 cells were harvested, washed with ice-cold phosphate-buffered saline (PBS), resuspended in 200 μL of binding buffer [10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-NaOH pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2], and incubated with 5 μL of annexin V conjugated with FITC for 10 minutes at room temperature in the dark. The samples were then washed with binding buffer, resuspended in PBS, counterstained with propidium iodide (PI), and analyzed on the Gallios flow cytometer and with the Kaluza analysis software. The annexin V−/PI+ cells were assumed to be necrotic, annexin V+/PI+ cells were considered late apoptotic, and annexin V+/PI− cells were assumed to be early apoptotic.
9. Preparation of cell lysates and immunoblot analysis
Whole-cell lysates were prepared as follows. Cells were washed twice with ice-cold PBS, then harvested by scraping into lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 μg/mL aprotinin, 10 μg/mL leupeptin, 20 mM β-glycerophosphate, and 2 mM sodium fluoride). Cell lysates were clarified by centrifugation at 15,000 ×g for 10 minutes at 4°C, after which lysate protein concentrations were determined with the BCA Protein Assay Reagent Kit (Pierce, Rockford, IL). For immunoblotting, whole-cell lysates were denatured in Laemmli sample buffer, then resolved by SDSpolyacrylamide gel electrophoresis. The proteins were transferred to a nitrocellulose membrane, after which immunoblotting analysis was performed using the appropriate primary antibodies. The immune complexes were visualized with an Amersham ECL Kit (GE Healthcare Life Sciences) in the dark and images were captured using the BioRad Chemi-Doc XRS+ instrument (Hercules, CA). Band intensity values were measured using the BioRad Image Lab software ver. 5.2.1 (BioRad Laboratories Inc.).
Results
1. TaqMan qRT-PCR confirmation of differentially regulated miRNAs
To determine which miRNAs are responsible for resistance to BRAF inhibitors, we examined changes in miRNA expression in three cell lines: BRAF inhibitor–sensitive A375P BRAF V600E cells and two BRAF inhibitor–resistant cell lines (A375P/Mdr and SK-MEL-2 BRAF-WT). The miRNAs with an at least two-fold change in expression between BRAF inhibitor–sensitive and –resistant cell lines were identified as differentially expressed. Averaged intensity measurements identified 43 miRNAs with expression that was consistently altered two-fold or more in two BRAF inhibitor-resistant cell lines, regardless of whether the resistance was intrinsic or acquired. Of these, 25 miRNAs were consistently upregulated and 18 were downregulated by more than two-fold.
To validate the results of the microarray analysis, qRT-PCR was conducted for the 22 selected miRNAs (10 upregulated and 12 downregulated miRNAs) using the TaqMan MicroRNA Assay (Table 1). Some discrepancies were detected when miRNA microarray data were collated with qRT-PCR–measured expression levels. Nevertheless, qRT-PCR analysis of five miRNAs showed excellent agreement with the microarray experiments (Fig. 1). We found miR-1246, miR-193b-3p, and miR-3617 to be consistently upregulated in the two BRAF inhibitor–resistant cell lines. In contrast, miR-17-3p and miR-92a-1 were downregulated in the two BRAF inhibitor–resistant cell lines.
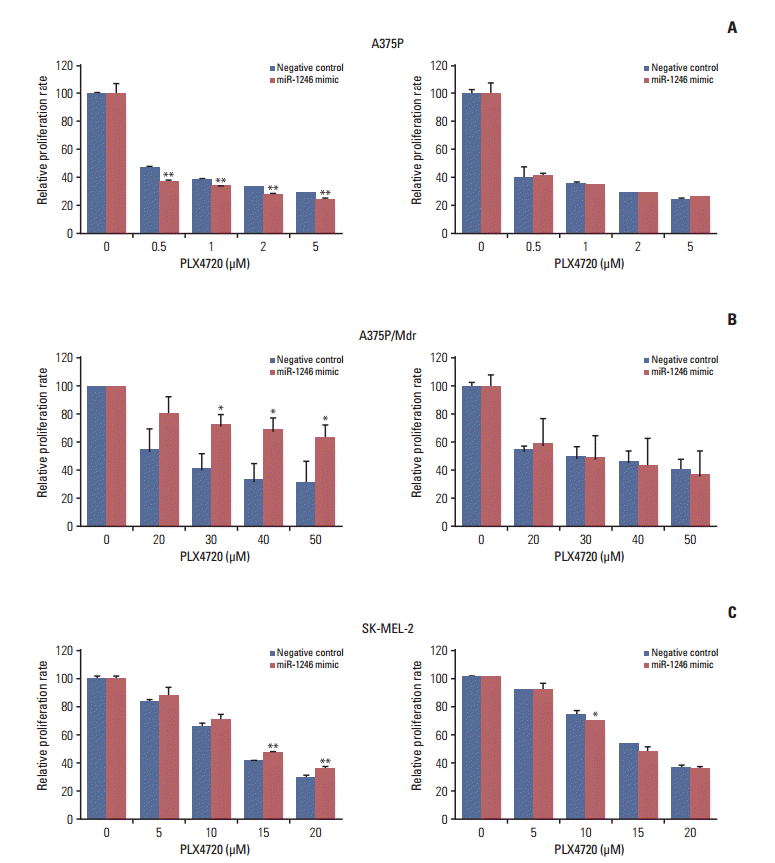
2. The miR-1246 mimic confers resistance to oncogenic BRAF inhibitors in A375P/Mdr cells
To explore the cellular functions of miRNAs associated with resistance to BRAF inhibitors, the effects of miRNAs on cell proliferation were determined after transient transfection (Fig. 2). Synthetic oligonucleotide miRNA mimics and inhibitors were transfected into three cell lines, and proliferation was quantified by a colorimetric assay. Of the five miRNAs tested (S1 Fig.), miR-1246 (which was upregulated in BRAF inhibitor–resistant cells) conferred strong additional PLX4720 resistance to A375P/Mdr cells (Fig. 2B), implying that miR-1246 upregulation is responsible for resistance to BRAF inhibition. In addition, in SK-MEL-2 cells (intrinsically resistant to BRAF inhibition), the difference in cell growth rate between mimic-control-transfected and miR-1246 mimic-transfected cells was statistically significant; however, the efficacy of miR-1246 was modest (Fig. 2C). MiR-1246 did not cause PLX4720 resistance in BRAF inhibitor–sensitive A375P cells. Moreover, miR-1246 inhibitors showed negligible effects on PLX4720-induced growth inhibition relative to the mimic control.
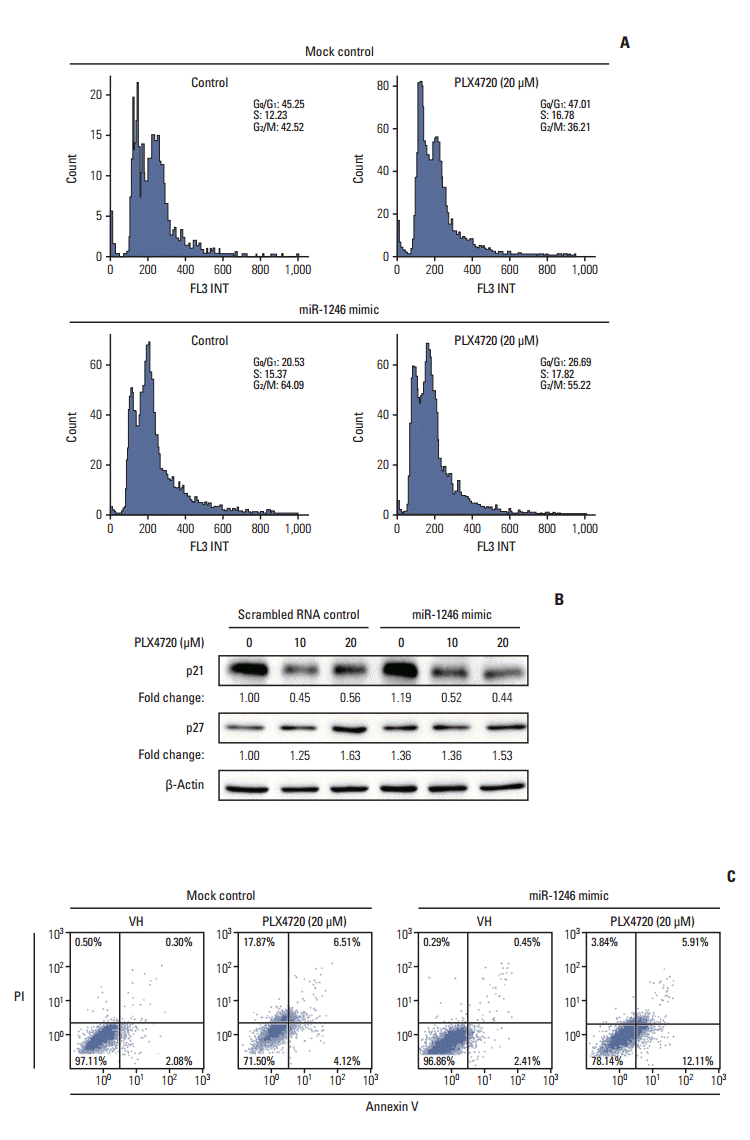
3. Effects of miR-1246 on PLX4720-induced G2/M arrest and apoptosis
To characterize the mechanism by which miR-1246 induces BRAF inhibitor resistance to A375P/Mdr cells, the effect of miR-1246 on cell cycle distribution was determined by flow cytometry. Cell cycle analysis showed a significant decrease in the cell population with 2N DNA content (G1: 45.25% in mimic-control-transfected population to 20.53% in population transfected with the miR-1246 mimic), whereas a marked increase was observed in the population with 4N DNA content (G2/M: 42.52% in mimic-control-transfected population to 64.09% in population transfected with the miR-1246 mimic) (Fig. 3A). The number of cells in the S phase was approximately unchanged (12.23% vs. 15.37%). We also investigated the expression of the cyclin-dependent kinase inhibitors (CKIs) p21Cip1 and p27Kip1 (Fig. 3B). We detected a remarkable decrease of p21Cip1 in A375P/Mdr cells after PLX4720 treatment. We previously found a relatively high background expression level of p21Cip1 in A375P/Mdr cells compared to A375P cells [15]. Conversely, PLX4720 treatment only moderately increased the levels of p27Kip1 in A375P/Mdr cells. However, miR-1246 had no effect on PLX4720-induced changes in the levels of p21Cip1 and p27Kip1, which implies that the miR-1246-mediated increase in G2/M phase was not mediated by CKIs.
Because chemotherapeutic agents can cause apoptosis after G2/M arrest, apoptotic cell death was assessed by staining with FITC-labeled annexin V and PI. The data presented in Fig. 3C revealed that the primary mechanism of cell death appeared to differ between miR-1246 mimic-transfected and mimic-control-transfected A375P/Mdr cells. For the miR-1246 mimic-transfected cells, PLX4720 resulted in the significant emergence of apoptotic cells, but not necrotic cells. In miR-1246 mimic-transfected cells, the necrosis/apoptosis ratio was 0.32. Conversely, among the mimic-control-transfected cells, the majority died via necrosis instead of apoptosis. The results showed the ratio of necrosis/apoptosis in mimic-control-transfected cells was 4.33. These data imply that the difference in sensitivity to PLX4720 between mimiccontrol-transfected and miR-1246 mimic-transfected cells may result from the difference in the proportion of the necrotic cell population.
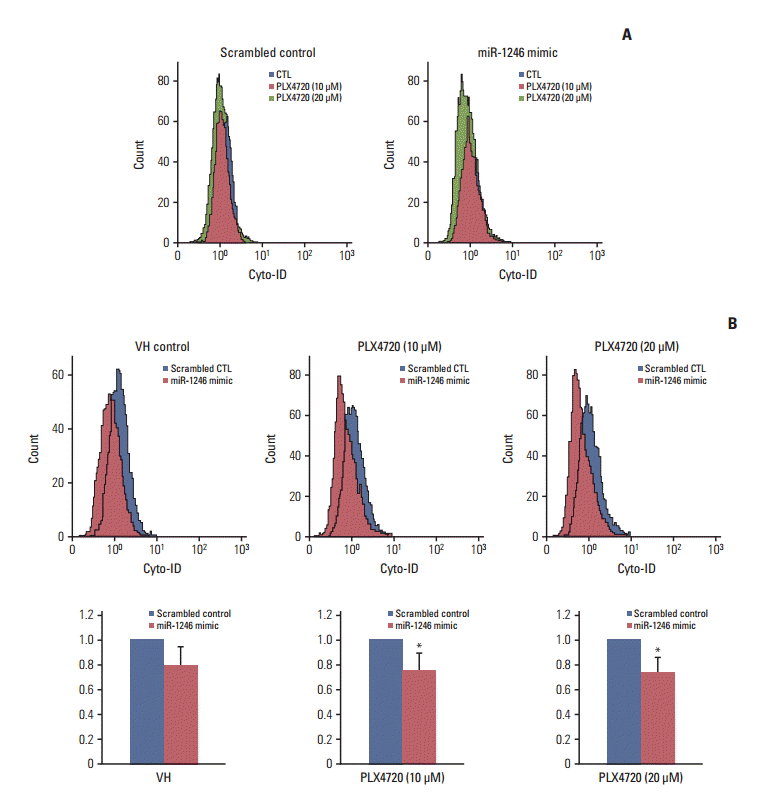
4. The role of autophagy in miR-1246-mediated resistance to the BRAF inhibitor
We previously reported that inhibitors of oncogenic BRAF offer an effective therapeutic strategy against A375P BRAF V600E melanoma by modulating autophagy [12,16]. Thus, we studied the role of autophagy in PLX4720-induced growth inhibition using miR-1246–transfected cells. The change in autophagy was examined by flow cytometry using Cyto-ID, a green fluorescent dye that accumulates in autophagic vacuoles [14]. This assay was recently demonstrated to be a rapid and quantitative method for monitoring of autophagy [17]. A representative flow cytometry histogram of Cyto-ID fluorescence is shown in Fig. 4A. Treatment with PLX4720 resulted in a dose-dependent decrease in autophagy of miR-1246 mimic-transfected cells; however, it did not affect autophagy of mimic-control-transfected cells. Fig. 4B shows that, when compared with mimic-controltransfected cells, PLX4720 significantly decreased Cyto-ID fluorescence intensity in miR-1246-transfected A375P/Mdr cells. These results imply that the miR-1246-induced inhibition of autophagy may contribute to the resistance to BRAF inhibitors.
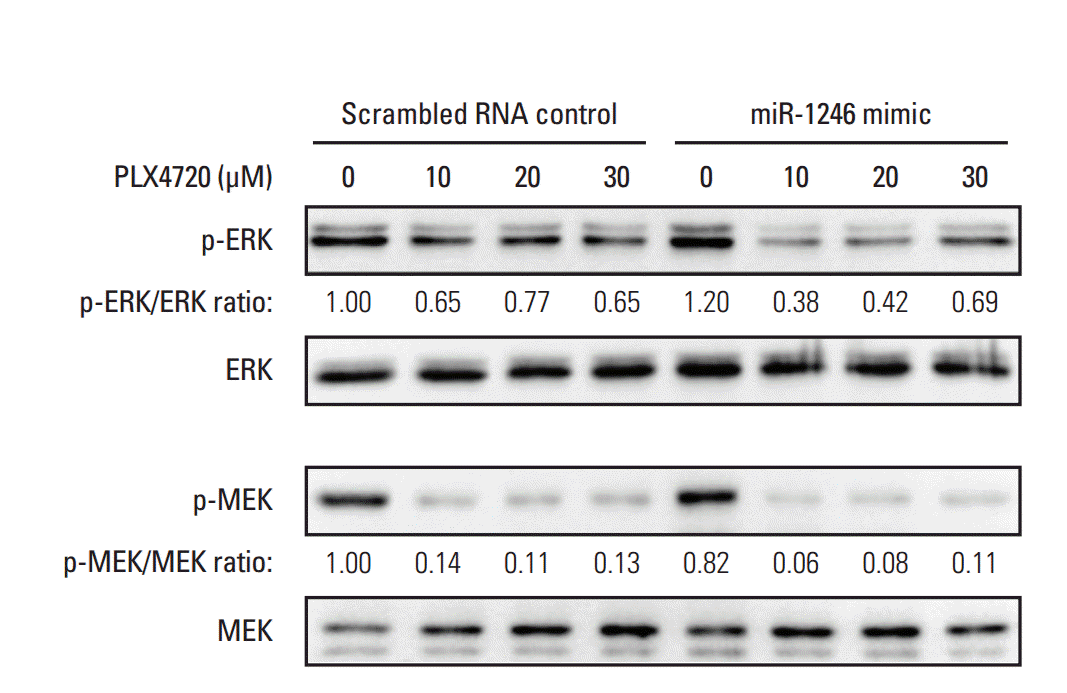
5. Effects of miR-1246 on MEK-ERK signaling
Because it is known that A375P/Mdr cells are resistant to BRAF inhibitor-induced ERK inhibition [6], we used immunoblotting to examine phospho-MEK and phospho-ERK levels in mimic-control-RNA–transfected and miR-1246–transfected A375P/Mdr cells after PLX4720 treatment (Fig. 5). As previously reported, p-ERK levels remained relatively elevated in mimic-control-transfected cells in the presence of PLX4720. Just as in BRAF inhibitor–sensitive A375P cells, marked downregulation of p-ERK was detected in miR-1246–transfected A375P/Mdr cells after PLX4720 treatment. Nonetheless, PLX4720 almost completely abrogated the phosphorylation of MEK, the upstream activator of ERK, in both cell types. These results suggest that the promotion of BRAF inhibitor resistance by miR-1246 was associated with lowered levels of p-ERK.
Discussion
The onset of acquired resistance limits clinical efficacy of BRAF inhibitors [4]. Although several studies have explored the roles of miRNAs in melanoma development and progression [10,18], little is known about the possible involvement of microRNAs in acquisition of BRAF inhibitor resistance. Here, we identified 43 miRNAs associated with resistance to BRAF inhibitors using a miRNA array platform. TaqMan quantitative RT-PCR (qRT-PCR) analyses showed that five of these miRNAs (miR-3617, miR-92a1, miR-1246, miR-193b-3p, and miR-17-3p) may be associated with resistance to the BRAF inhibitor, confirming the change in expression of these miRNAs in two BRAF inhibitor–resistant cell lines. These five miRNAs have not been extensively studied in terms of resistance to BRAF inhibitors; however, some play oncogenic or tumor suppressor roles in cancers. miR-92a and miR-17-3p are upregulated in tumor cells and associated with increased proliferation and tumorigenesis [19,20]. Among the best studied is miR-1246, which was first identified during sequencing of human stem cells in 2008 [21]. Nonetheless, there is an ongoing debate regarding whether miR-1246 acts as an oncomicroRNA or tumor suppressor. Although the expression of miR-1246 has been reported to be negatively correlated with cancer progression [22], many other studies revealed that miR-1246 can enhance cell migration and invasion in cancer cells [23], supporting the oncogenic role of miR-1246. In particular, several reports on miR-1246 have shown that miR-1246 promotes tumorigenesis by targeting cytoplasmic polyadenylation element-binding protein 4 [24] and thrombospondin 2 (THBS2) [25].
Based on microarray data, the average level of miR-1246 was found to be over threefold higher in two BRAF inhibitorresistant cells than in BRAF inhibitor–sensitive cells. A cell growth assay revealed that the miR-1246 mimic confers the strongest resistance to the BRAF inhibitor (PLX4720) in A375P/Mdr cells. In addition, miR-1246 mimic also significantly reduced the antiproliferative effects of PLX4720 in SK-MEL-2 cells, another cell line resistant to BRAF inhibitors; however, the efficacy of miR-1246 was modest. Notably, our miR-1246 mimic failed to reverse the growth inhibition by PLX4720 in BRAF inhibitor–sensitive A375P cells. Thus, these results indicate that a target gene of miR-1246 may be upregulated in cells with acquired resistance to BRAF inhibitors. In agreement with our results suggesting that miR-1246 expression is associated with resistance to anti-BRAF therapy, the in vitro drug sensitivity of pancreatic cancer cells is known to be altered depending on miR-1246 expression [26]. Nevertheless, the exact mechanism of action of miR-1246 in drug resistance is not completely understood. Our previous study revealed that treatment with a BRAF inhibitor leads to a dose-dependent apoptotic changes in BRAF inhibitor-sensitive A375P cells [3]. In the present study, our flow cytometry results clearly showed necrosis instead of apoptosis in BRAF inhibitor-resistant A375P/Mdr cells based on their being 17.87% PI-positive annexin V–negative cells versus only 4.12% PI-negative annexin V–positive cells. One possible mechanism behind the higher rate of necrosis in BRAF inhibitor–resistant A375P/Mdr cells may be upregulation of antiapoptotic pathways in A375P/Mdr cells, which may shift the cells to necrosis during PLX4720 treatment. Conversely, miR-1246 mimic transfection induced very little necrosis, but a moderate degree of apoptosis in A375P/Mdr cells. These results indicate that resistance to PLX4720 in miR-1246 mimic-transfected cells is predominantly due to a reduction in necrosis concomitant with a moderate increase in apoptosis. It has been reported that miR-1246 can induce p53-dependent apoptosis in response to DNA damage [27].
Nevertheless, autophagy has two seemingly contradictory effects on tumor development. Increasing evidence indicates that autophagy can either promote cell survival at certain stages or cell death at other stages [28]. In our study, PLX4720 produced a significant decrease of autophagy in miR-1246–transfected cells; however, it had no inhibitory effect in mimic-control-transfected cells, implying that the miR-1246–induced inhibition of autophagy may contribute to resistance to BRAF inhibitors. In fact, G2/M arrest is associated with a survival mechanism for cancer cells as a way to escape cell death induced by anticancer therapy [29]. In the present study, we found that PLX4720 caused much more G2/M arrest in A375P/Mdr cells transfected with the miR-1246 mimic than that seen in mimic-control-RNA-transfected cells. Thus, our results indicate that miR-1246 increases the cell population in the G2/M phase through autophagy inhibition and promotes escape from cell death by necrosis and apoptosis in response to PLX4720.
The continued activation of the MAPK pathway observed in the presence of PLX4720 is believed to be a major cause of BRAF resistance in BRAF inhibitor–resistant cells. It has been reported that activation of ERK leads to negative feedback regulation of MEK [30]. Our previous results also showed that long-term treatment with BRAF inhibitors significantly downregulated Spry2 in BRAF V600E–positive cell lines, and that this change is accompanied by rebound activation of the MAPK pathway [6]. As expected, the phospho-ERK levels were relatively unresponsive to PLX4720 in mimic-controltransfected A375P/Mdr cells. Conversely, a decrease in the amount of p-ERK was noted in miR-1246 mimic-transfected A375P/Mdr cells, suggesting that the promotion of BRAF inhibitor resistance by miR-1246 may be associated with lowered levels of p-ERK. Interestingly, in miR-1246 mimic-transfected A375P/Mdr cells, PLX4720 induced the phosphorylation of ERK in a slight dose-dependent manner up to 30 μM. We previously reported that PLX4720 induced paradoxical phosphorylation of ERK in SK-MEL-2 cells that showed a much lower level of p-ERK compared to A375P cells with elevated levels of p-ERK [3]. Thus, in this study, we cannot exclude the possibility that, as in SK-MEL-2 cells, PLX4720 induced the paradoxical activation of ERK in A375P/Mdr cells in which p-ERK was markedly downregulated by miR-1246. These data point to another mechanism behind the dysregulation of the MAPK pathway that may contribute to the profound resistance associated with current BRAF-targeted therapies.
Taken together, our results indicate that miR-1246 may be a potential therapeutic target in melanoma with acquired resistance to BRAF inhibitors. Nonetheless, the mechanism underlying the miR-1246–induced resistance to the BRAF inhibitor is unknown. Thus, a better understanding of miR-1246 regulation should advance our knowledge of the molecular mechanisms behind resistance to chemotherapy. Further research investigating a miR-1246 knockout model is needed to identify the potential target genes of miR-1246 and fully evaluate the functional impact and importance of miR-1246 in melanoma cells.