INTRODUCTION
Neuroblastoma is the most common extracranial childhood solid tumor and is associated with marked clinical, histological, and genetic heterogeneity. Most often, progression of the clinical course results in a poor response to conventional treatments. The prognosis of neuroblastoma is reliant on genetic alterations of the cancer cells, which may include non-random deletions, rearrangements, or amplification within the chromosome. Among these alterations, MYCN amplification appears to be the most influential. Because over 60% of patients die as the result of aggressive progression, it is necessary to develop and use therapeutic agents in treatments for neuroblastoma, regardless of the heterogeneous nature and genetic alterations of the neuroblastoma even in the case of MYCN amplification. In addition, the inhibition of MYCN gene expression in MYCN-amplified cells induces apoptosis and suppresses cell growth (1), which suggests its potential as a therapeutic target for the treatment of aggressive neuroblastomas by small interfering RNA (siRNA) to MYCN.
However, MYCN expression might enhance the anti-tumor effects of various drug therapies because MYCN expression induces the transition from the G1-S to G2/M phase (2). Chemotherapeutic drugs exploit several biological mechanisms for the targeting of cancer cells including mitotic inhibition, microtubule formation inhibition, or DNA alkylation. All these anti-cancer activities of the drugs generally target cancer cells in the M or S-phases of the cell cycle. Therefore, it is necessary to assess the effects of N-Myc knockdown by siRNA on cell cycles in combination with Imatinib Mesylate (Gleevec, STI-571), which is considered a promising new therapeutic drug for the treatment of aggressive neuroblastomas, and which may also be administered in combination with other chemotherapeutic agents that target the M or S-phase of cells.
STI-571 is a selective inhibitor of Bcr-Abl tyrosine kinase in addition to other tyrosine kinases; e.g. the PDGF receptor (PDGFR) and c-Kit (3). STI-571 is currently employed for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST), and its potential uses for the treatment of other c-Kit-positive malignancies including seminoma and acute myeloid leukemia are currently being investigated (4). Although c-Kit is preferentially expressed in MYCN-amplified neuroblastoma and STI-571 inhibited cell proliferation in vitro (5), the effect of STI-571 on the MYCN-amplified neuroblastoma cells had not been clearly shown.
In this study, we attempted to determine the effects of STI-571 on the growth of neuroblastoma cells in vitro, depending on the state of MYCN amplification and expression, using SK-N-BE(2) cells. For the knockdown of N-Myc expression, we utilized MYCN siRNA. We then assessed the cytotoxicity and cell cycle distribution in STI-571-treated SK-N-BE(2) cells.
MATERIALS AND METHODS
1) Cell culture and chemicals
STI-571 was generously provided by Novartis (Basel, Switzerland). It was prepared as a 10 mM stock solution in a sterile solution in dimethylsulfoxide (DMSO). The neuroblastoma cell line SK-N-BE(2) (CRL-2271, ATCC) was cultured in RPMI1640 medium supplemented with heat-inactivated 10% fetal bovine serum and antibiotics.
2) siRNA transfection
siRNA against MYCN (siMYCN, siGENOME SMARTpool M-003913-01-0005) (1) was purchased from Dharmacon (Lafayette, CO), and the non-targeting control was acquired from Bioneer (Daejeon). Transfection was conducted with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), in accordance with the manufacturer's recommendations. The cells were seeded on 6-well plates for protein extraction and cell cycle analysis.
3) Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from siRNA-transfected cells using TRIZOL reagent (Invitrogen), and RT-PCR was conducted in order to confirm cellular gene expression. The primer sequences were as follows: MYCN forward (5'-ACC ACA AGG CCC TCA GTA CC-3'), MYCN reverse (5'-GTG CAT CCT CAC TCT CCA CG-3'), GAPDH forward (5'-GTC TTC TCC ACC ATG GAG AA-3'), and GAPDH reverse (5'-CAT GCC AGT GAG CTT CCC GTT CA-3'). PCR conditions were denaturation for 5 min at 94℃, 30 cycles of amplification for 1 min each at 94℃, 1 min at 55℃, and a final extension for 1 min at 72℃.
4) AnnexinV and propidium iodide staining
Cells (5×105 cells/well) were treated for 72 h with STI-571 after siRNA transfection. The cells were detached with trypsin and washed twice in cold phosphate buffered saline (PBS), after which the cells were resuspended in 1× Binding Buffer (10 mM HEPES, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2) at a concentration of 1×106 cells/ml. The cells were stained with AnnexinV and PI for 15 min at room temperature in darkness. For the cell cycle analysis, cells were fixed with 70% ethanol and the cellular DNA was stained by incubating the cells in 500 ul of PBS containing 50 ug/ml PI, 5 mM EDTA, and 1 mg/ml of RNase for 30 min at room temperature. After staining, the cells were analyzed on a FACSCalibur system (BD Biosciences, San Jose, CA) with CellQuest software (BD Biosciences) and ModFitLT software (Verity Software House, http://www.vsh.com).
5) MTT assay
For the 3-(4,5-dimethylthizol-2-yl)2,5-diphenyl tetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO) assay, cells were plated onto 96-well microtiter plates at a density of 5×103/150 ul in fresh medium, then treated with arsenic trioxide and/or STI-571. After 72 h, 20 ul of MTT (5 mg/ml in PBS) was added to each well and the plates were returned to the incubator for an additional 4 h. At the end of the incubation period, the supernatants were discarded by suction and 200 ul of DMSO were added to all wells in order to dissolve the dark blue formazan crystals. The plates were subsequently covered with aluminum foil, gently shaken, and read at a wavelength of 570 nm.
6) Immunoblot procedure
Cells were lysed in a buffer containing 150 mM NaCl, 1% NP-40, 0.5% deoxychloine (DOC), 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 7.5), 2 mM sodium orthovanadate, 2 µg/ml phenylmethylsulfonyl fluoride (PMSF), and 2 µg/ml of aprotinin. The lysates were sonicated with an ultrasonic homogenizer (Cole-Parmer, Chicago, IL) and centrifuged for 10 min at 11,000 rpm at 4℃. The clear lysates were collected and the protein concentrations of the lysates were determined using a BCA kit (Pierce Chemical Co., Rockford, IL). The protein was then electrophoretically separated (5 µg per lane) by 10% SDS-polyacrylamide gel electrophoresis. The antibodies utilized were anti-MYCN Ab (Cell Signaling Technology, Beverley, MA), and anti-beta-actin Ab (Sigma-Aldrich). Anti-cyclin E Ab (C-19), anti-cyclin B1 Ab (H-433), anti-cyclin D1 Ab (C-20), anti-p27 Ab (C-19), anti-CDK1 Ab (clone 17), anti- CDK4 Ab (H-22), and anti- CDK2 Ab (H-298) were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies were purchased from BioRad (Hercules, CA).
RESULTS
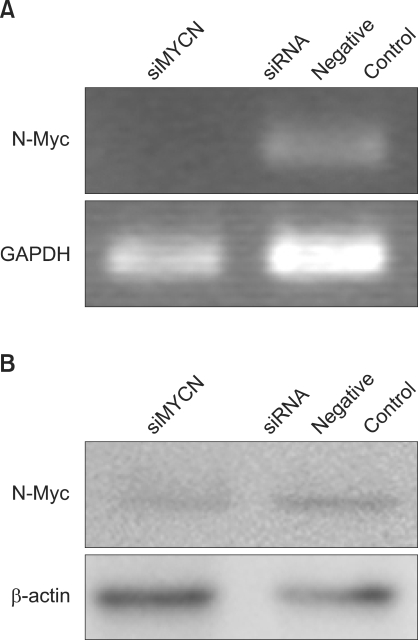
1) Influence of siRNA on N-Myc expression
To characterize the effects of N-Myc knockdown on STI-571 treatment, MYCN knockdown was induced via siMYCN. Use of siMYCN and negative controls revealed decreased N-Myc protein expression after siRNA treatment (Fig. 1).
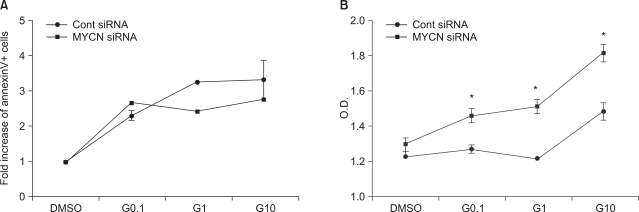
2) Influence of siMYCN on cytotoxic effects
To clarify the effects of N-myc knockdown on STI-571 and arsenic trioxide treatment, cells were transfected with siMYNC or siRNA negative controls and staineded with AnnexinV for the detection of apoptotic cells. Although the fraction of apoptotic SK-N-BE(2) cells was reduced after the administration of STI-571 (Fig. 2A), siMYCN transfected cells induced a lower level of apoptosis in the STI-571-treated cells than was seen with control siRNA-transfected SK-N-BE(2) cells (Fig. 2B).
In the MTT assay, control siRNA transfected cells induced less proliferation of SK-N-BE(2) cells after STI-571 treatment (Fig. 2B) than was observed for siMYCN-transfected cells. This indicates that N-Myc knockdown did not render the neuroblastoma cells more sensitive to STI-571 treatment.
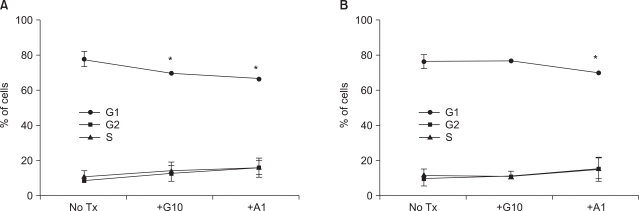
3) Influence of MYCN knockdown on arrest of SK-N-BE(2) cells in the G0/G1 phase after STI-571 treatment
siMYCN-treated SK-N-BE(2) cells displayed a lower level of apoptosis and a higher level of proliferation after STI-571 treatment. Therefore, we elected to analyze the cell cycle after STI-571 and arsenic trioxide treatments. Because the arsenic trioxide-treated cells displayed DNA damage and cell cycle arrest at the G0 or G2/M phase in the neuroblastoma cell lines, we compared the results of these treated cells with those of STI-571-treated cells. In the control siRNA-transfected cells, the proportion of G0/G1 cells was 80.3%. After 72 h of treatment with 10 uM of STI-571 or 1 uM of arsenic trioxide the proportion of G0/G1 cells was reduced up to 72.02% and 69.39%, respectively (Fig. 3A). In siMYNC-transfected SK-N-BE(2) cells, the proportion of G0/G1 cells displayed no significant change after treatment with 10 uM of STI-571 (Fig. 3B).
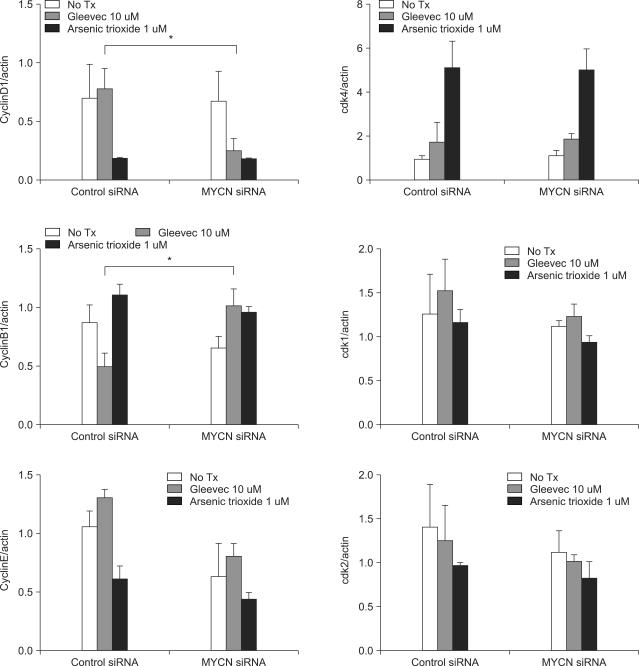
Because the siMYCN-transfected cells were arrested in the G0/G1 phase after STI-571 treatment, we needed to determine the levels of cyclin and CDK proteins after 72 h of treatment. As shown in Fig. 4, 5, cyclin D expression was reduced and cyclin B1 expression was increased in siMYCN-transfected SK-N-BE(2) cells as compared to the control siRNA-transfected cells. As cyclin D levels have been shown to increase in the mid-G1 phase of the cell cycle, the reduction in the cyclin D level may have caused an increase in the proportion of cells in the G1 cycle. In addition, increases in cyclin B levels in the G2 and M phases corresponded to progression to the G1 phase as well. The levels of cyclin E, CDK1, CDK2, and CDK4 did not differ between the two groups.
DISCUSSION
In this study, we demonstrate that the gene knockdown of N-Myc via RNA interference in SK-N-BE(2) cells inhibits the apoptotic effects associated with STI-571 exposure, and that the N-Myc knockdown cells remain in the G0/G1 cell cycle phase after STI-571 treatment.
The Myc protein is a member of the family of basic-helix-loop-helix-leucine zipper transcription factors (6). MYCN amplification occurs in approximately 25% of neuroblastoma cases (7), and is associated with cancer progression and treatment failure (8). Although the linkage between the mechanism of N-Myc overexpression and more aggressive neuroblastoma phenotypes remains to be elucidated, MYCN clearly induces cell growth via transcriptional activation of the target genes ODC, MCM7, MDM2, MDR1, or PAX3, and is involved in the progression of the G1-S phase, bypassing the G1 checkpoint (9). As MYCN-expressing cancer cells often evidence defects in the apoptotic pathway, the inhibition of MYCN expression may prove to be a useful target for neuroblastoma treatment. In service of such objectives, RNA interference can be employed for the knockdown of specific proteins in target cells. Appropriately, siRNA technique-directed MYCN has been recently highlighted as a potential new therapeutic modality for the treatment of aggressive neuroblastoma (1).
If the siRNA technique for MYCN knock-down were adopted for the treatment of the advanced form of neuroblastoma, the possible effect of combination treatment with other chemotherapeutics should be considered, because most chemotherapeutics target proliferating cancer cells via mitotic inhibition, thereby preventing the formation of microtubules, inhibiting disassembly into tubulin, and inhibiting topoisomerase and DNA alkylation. This means, for combination treatment of the neuroblastoma cells must be arrested in the G2-M or S phases. Moreover, anti-tumor drugs in MYCN-on cells result in G2-M and increase apoptosis, but MYCN-off cells are arrested in G1, and exhibit a delayed onset of apoptosis (2). In this study, MYCN knockdown itself caused no significant difference in the fraction of G0/G1 phase cells as compared to control siRNA-transfected cells after arsenic trioxide treatment (Fig. 3). MYCN knockdown by RNAi, however, affected the progression of the cell cycle after STI-571 treatment (Fig. 3B).
STI-571 treatment applied to neuroblastoma cells has been targeted to c-Kit and PDGFR-α and -β (10). In particular, c-Kit and its ligand, stem cell factor, areexpressed in neuroblastoma cells, e.g., SK-N-BE(2) cells (11), in addition to primary tumors (4). Moreover, c-kit is preferentially expressed in MYCN-amplifying neuroblastoma and inhibits STI-571-mediated proliferation (5). Presently, SK-N-BE(2) cells increased the fraction of apoptotic cells after exposure to STI-571, but this was less true in siMYCN transfected cells (Fig. 2A). This result implies that siMYCN treatment may interfere with the STI-571 induced anti-cancer effect on neuroblastoma cells.
CONCLUSION
In summary, MYCN knockdown via the siRNA method coupled with STI-571 treatment induces a lesser degree of apoptosis and arrested G0/G1 phase in an SK-N-BE(2) neuroblastoma cell line. Therefore, chemotherapeutic drugs targeting S or G2-M phase may prove ineffective when applied to cells arrested in the G0/1 cycle due to MYCN knockdown or STI-571 treatment.