Introduction
Tumor suppressor TP53 gene mutations are frequently detected in most human cancers [1]. TP53 can induce DNA repair, terminal differentiation, cell growth arrest, and apoptosis in response to potential oncogenic cellular stress such as DNA damage [2-4]. Therefore, TP53 loss-of-function mutations inhibit cell growth arrest and/or apoptosis, leading to initiation of carcinogenesis.
Many previous studies on breast cancer have reported that TP53 loss-of-function results in mammary carcinoma [5,6]. Li-Fraumeni syndrome, a hereditary tumor syndrome generating familial breast cancer and other neoplasms, is caused by germline TP53 mutations [7]. This report showed that germ line TP53 mutation resulted in a loss of p53 expression and most subsequent studies on TP53 have concerned the use of TP53 functional loss as a prognostic or predictive marker in breast cancer [8]. However, even though TP53 is a tumor suppressor gene, three quarters of TP53 mutations are missense substitutions rather than truncation/deletion mutations, such as nonsense or frameshift mutations [9]. Indeed, recent studies showed that missense mutations have functionally heterogeneous phenotypes and have different transcriptional activities depending on the locus of single nucleotide substitutions [10], and some mutant loci exhibit gain-of-function properties [11,12]. The most frequent missense mutants observed in breast cancer encode R175H, R248Q, R273H, and Y200C mutation. Among these four hot spots, R175H and R273H are gain-of-function mutations [9].
The Cancer Genome Atlas (TCGA) project for breast cancer recently showed that the most frequent genetic alterations are observed in TP53 [13]. TP53 mutations are more frequently observed in HER2-enriched and basal-like subtypes (72% and 80%, respectively) than in luminal A and B subtypes (12% and 29%, respectively). Using pathway analysis, the TCGA project found that the p53 pathway was intact in luminal A type cancer, in contrast with the loss of TP53 observed in basal-like tumors. However, not all basal-like tumors with TP53 mutations have a disrupted p53 pathway.
Many researchers have tried to determine the impact of TP53 mutation on breast cancer patient prognosis. Some previous studies reported that mutant TP53 indicated poor prognosis in primary breast cancer [8,14] and other research showed that an expression signature of TP53 status precisely predicted prognosis of breast cancer patients, unlike TP53 mutant status [15]. In addition, another previous study reported that only missense mutations in the DNA-binding domain of TP53 affect clinical outcome [16].
However, no studies have clarified the value of TP53 status as a prognostic or predictive marker in triple-negative breast cancer (TNBC), a subtype that most commonly has TP53 mutations [13]. Accordingly, we analyzed the relationship between TP53 status and clinical outcome of TNBC patients.
Materials and Methods
1. Patients
This study was a retrospective analysis of the clinical records of patients with invasive breast cancer who received adjuvant chemotherapy after curative surgery at Samsung Medical Center between 2000 and 2004. Women diagnosed with breast cancer stage I to IIIC by diagnostic examinations (breast magnetic resonance imaging, abdominal computed tomography [CT] scan, bone scan, and/or positron emission tomography–CT scans if indicated) were included.
This study was reviewed and approved by the Institutional Review Board (IRB) of Samsung Medical Center, Seoul, Korea (IRB No. 2012-08-065) with an informed consent waiver, due to the use of archival tissues with retrospective clinical data.
2. Immunohistochemical staining
Two experienced pathologists reviewed all pathology specimens to determine the following tumor characteristics: histological and nuclear grade, tumor size, lymphovascular invasion, tumor embolism, and immunohistochemical (IHC) staining for estrogen receptor (ER), progesterone receptor (PgR), HER2, and p53.
Triple negativity was defined as a lack of ER, PgR, and HER2 expression. Lack of ER and PgR expression was defined using Allred scores ranging from 0 to 2 based on IHC using antibodies to ER (Immunotech, Marseilles, France) and PgR (Novocastra Laboratories Ltd., Newcastle upon Tyne, UK). HER2 status was evaluated using a specific antibody (DAKO, Glostrup, Denmark) and/or silver in situ hybridization. Grades 0 and 1 for HER2, as assessed by IHC, were defined as a negative result. Amplification of HER2 was confirmed by fluorescence in situ hybridization if HER2 was rated as 2+ by IHC. IHC for p53 was performed using an anti-human monoclonal p53 antibody (Invitrogen, New York, NY) at 1:4,000 dilution with an autoimmunostainer (Leica Bond Polymer Refine detection kit/Leica Bond-Max staining system, Leica Microsystems, Wetzlar, Germany). In assessing the positivity of immunostaining for each section, nuclear staining > 10% was regarded as positive.
Our pathologists also reviewed all core biopsies from referring institutions, including IHC performed at the time of initial referral, and findings for all surgical specimens without knowledge of NanoString results or treatment outcome.
3. DNA extraction
Tumors consisting of over 75% malignant cells were dissected under microscopy from 4-mm unstained sections by comparison with a hematoxylin and eosin (H&E) stained slide, and genomic DNA was extracted using a Qiagen DNA FFPE Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After extraction, concentration as well as 260/280 and 260/230 nm ratios were measured by spectrophotometry (ND1000, NanoDrop Technologies, Thermo Fisher Scientific, Waltham, MA). Each sample was then quantified using a Qubit fluorometer (Life Technologies, Carlsbad, CA). Genomic DNA with a total yield > 10 ng was used for library preparation.
4. RNA extraction
Areas containing representative invasive breast carcinoma were outlined on the slide. Total RNA was extracted from two to four sections of 4-μm-thick formalin-fixed, paraffinembedded sections. Non-tumor elements were removed by manual microdissection before transfer to the extraction tube, guided by H&E stained slides. Total RNA was then extracted using a High Pure RNA Paraffin kit (Roche Diagnostic, Mannheim, Germany). RNA yield and purity were assessed using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE). One sample with less than 50 ng/μL of total RNA concentration even after concentration with a SpeedVac concentrator (Thermo Scientific, Waltham, MA) was excluded from downstream analysis, because 200 ng of input RNA in a 5 μL volume was required for hybridization with 20 μL of the probe set mastermix.
5. nCounter expression assay (NanoString)
The level of gene expression was measured using the NanoString nCounter Analysis System (NanoString Technologies, Seattle, WA). The system measures the relative abundance of each mRNA transcript of interest using a multiplexed hybridization assay and digital readouts of fluorescent barcoded probes that are hybridized to each transcript [17]. An nCounter CodeSet (NanoString Technologies) containing a biotinylated capture probe for the TP53 gene and five housekeeping genes and reporter probes attached to color-barcode tags, according to the nCounter code-set design, was hybridized in solution to 200 ng of total RNA for 18 hours at 65°C according to the manufacturer’s instructions. Hybridized samples were loaded into the nCounter Prep Station for post-hybridization processing. On the deck of the Prep Station, hybridized samples were purified and immobilized in a sample cartridge for data collection followed by quantification of target mRNA in each sample using the nCounter Digital Analyzer. Quantified expression data were analyzed using NanoString nSolver analysis software. After performing image quality control using a predefined cutoff value, outlier samples were excluded using a normalization factor based on the sum of positive control counts greater than 3-fold. The counts of the probes were then normalized using the geometric mean of the five housekeeping genes. Accordingly, expression level means normalized mRNA transcript counts per 200 ng of total RNA extracted from tumor tissue. There is no unit of gene expression level measured by nCounter expression assay.
6. Next generation sequencing using Ion Torrent AmpliSeq Cancer Panel v2
Using the Ion Torrent Personal Genome Machine (Ion PGM, Life Technologies) Cancer Panel v2 (Supplemental Table 1), 2,855 loci from 50 cancer-related genes were sequenced for identification of genetic mutations. Libraries were constructed using the Ion AmpliSeq Panels pool (Life Technologies) with 10 ng of DNA sample per pool. The amplicons were then ligated to Ion Xpress Barcode Adapters and purified. Next, multiplexed barcoded libraries were enriched by clonal amplification using emulsion polymerase chain reaction on Ion Sphere particles (Ion PGM Template OT2 200 Kit, Life Technologies) and loaded onto an Ion 316 chip. Massive parallel sequencing was performed on the Ion PGM using the Ion PGM Sequencing 200 kit v2.
The primary filtering process was performed using Torrent Suite v3.6.0 and Ion Torrent Variant Caller v3.6 software. The pipeline included signaling processing, base calling, quality score assignment, adapter trimming, read alignment to 19 human genome references, mapping quality control, coverage analysis, and variant calling. For variant detection, a minimum coverage of 100 reads was achieved, and at least 5% of mutant reads were selected for variants. Variant calls were further analyzed using the ANNOVAR software tool, which includes variant filtering and annotation using the COSMIC database, dbSNP build 137, and information on amino acid change.
7. Bioinformatics and statistical analysis for AmpliSeq and nCounter assay
Variant calls from Ion AmpliSeq were further evaluated to reduce potential false-positives. Coverage (> 100×) and quality score (> 30) were considered as filtering criteria. In addition, a minimum threshold of mutant allele fraction was taken into account for confirming variants as real: > 1% for mutations with low allele fraction and > 10% for polymorphism. For statistical analysis of final variants, read alignments were manually examined using the Integrative Genomic Viewer (http://www.broadinstitute.org/igv/). Korean-specific germline variants in 50 genes were discarded via manual review using a Korean genome database [18]. Among variants satisfying the filtering criteria described above, variants causing amino acid change and frameshift were finally chosen for statistical analysis. The Fisher exact test was used for mutations and polymorphic variants separately for discovery of variants enriched in patients with favorable outcomes, and p-values less than 0.05 were considered significantly different.
8. Statistical analysis
Differences in clinicopathologic characteristics were analyzed using Student’s t tests. Distant recurrence-free survival (DRFS) was defined as the elapsed time from the date of curative surgery to the detection of distant recurrence. DRFS was analyzed using the Kaplan-Meier method. Two-tailed p-values < 0.05 were considered statistically significant, and IBM SPSS Statistics ver. 21 (IBM Co., Armonk, NY) was used for analysis of all data.
Results
1. Genetic mutation profile
In total, 203 TNBC patients who underwent curative surgery and received adjuvant chemotherapy were enrolled. Of the 203 patients, nCounter expression assay and targeted DNA sequencing were performed on 174 patients. Samples from 29 patients did not undergo next generation sequencing due to DNA extraction failure (Supplementary Fig. S1).
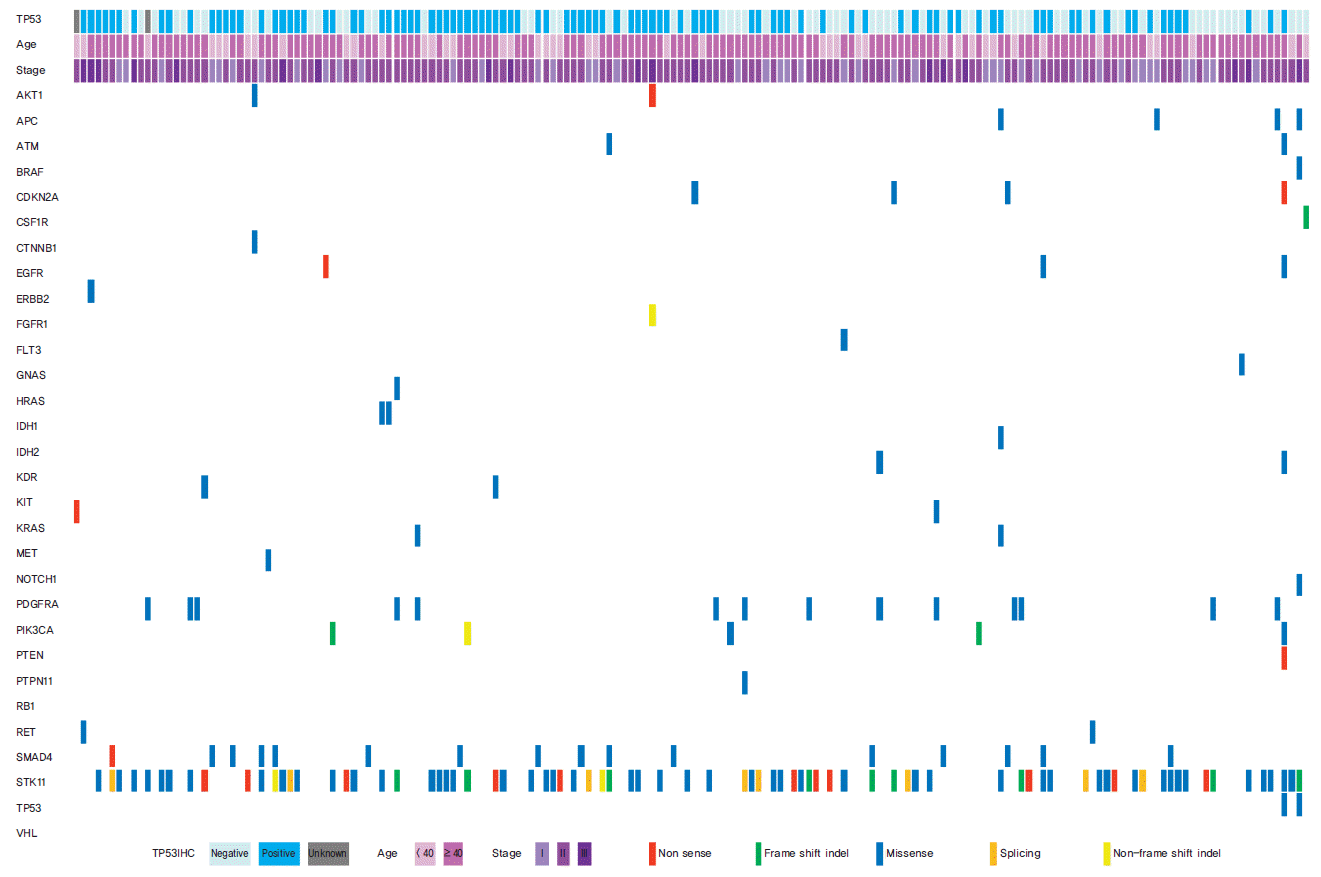
Of the 50 cancer-related genes examined (Supplementary Table 1), TP53 gene mutations were the most frequently found in TNBC patients (77 of 174 patients, 44.3%) (Table 1, Fig. 1). The second most frequent mutant gene was STK11 (9.6%), and the third was PIK3CA (9.0%). PTEN mutation was observed in four patients (2.6%), and mutations in the other 46 genes were detected in less than 2% of patients (Fig. 1).
2. Baseline characteristics including TP53 status
The characteristics of the 174 patients are shown in Table 1. p53 IHC showed that 103 patients (58.0%) showed p53 expression, while 69 patients (42.0%) had a loss of p53 expression. Two patients lacked any information on p53 expression. In mutation analysis, 77 patients (44.3%) had TP53 mutations; missense mutations were detected in 49 patients (63.6%), nonsense mutations in 20 patients (26.0%), and frameshift or splicing mutations of TP53 in eight patients (10.4%). The expression profile of the TP53 gene is also shown in Table 1. For further survival analysis, we set the median expression score of TP53 as the cut-off value to divide patients into two groups, lower versus higher expression.
We performed further analysis of the correlation between TP53 mutation and p53 expression status. Compared to patients with wild type TP53, patients with missense mutations in TP53 had higher protein expression, which contrasts with expression in patients with nonsense/frameshift or splicing mutations of TP53 (positivity of p53 IHC: wild type vs. missense vs. nonsense/frameshift/splicing [truncation/deletion] mutation, 53.6% vs. 89.8% vs. 25.0%, respectively; p < 0.001) (Table 2, Fig. 2A). In addition, expression levels of p53 were also influenced by TP53 mutation status (p53 expression level [median]: wild type vs. missense vs. truncation/deletion mutation, 207.36±132.73 vs. 339.61±143.21 vs. 99.53±99.57, respectively; p < 0.001) (Table 2, Fig. 2B). In terms of mRNA and protein expression, a positive correlation was observed between mRNA and protein expression (p53 expression level [median]: no expression vs. expression vs. unknown, 120.82±69.33 vs. 315.13±133.56 vs. 413.05±199.41, respectively; p < 0.001) (Fig. 2C).
3. Impact of baseline characteristics, including TP53 status, on patient clinical outcome
The 5-year DRFS (5Y DRFS) rate in patients with stage I or IIA disease was 88.4% and 91.9%, respectively, compared to 75.8%, 64.8%, and 33.0% in patients with stage IIB, IIIA, and IIIC, respectively (p < 0.001) (Table 1).
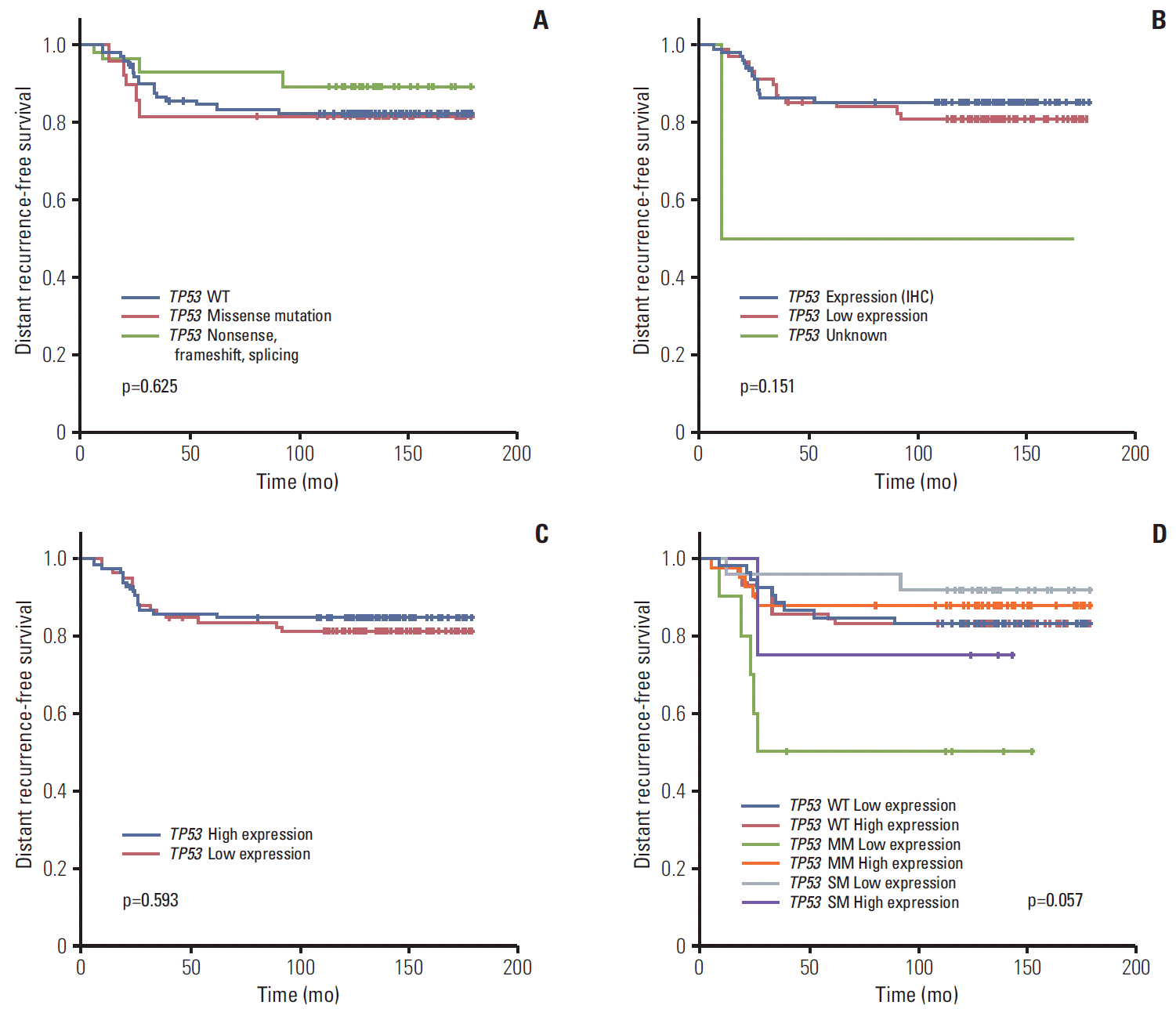
Neither TP53 mutation, p53 protein, or TP53 RNA expression status affected patient prognosis (5Y DRFS: wild type vs. missense vs. truncation/deletion mutation, 82.5% vs. 81.6% vs. 89.3%, respectively; p=0.625 in mutation profile; positive vs. negative, 85.4% vs. 81.2%, respectively; p=0.151 in IHC; low vs. high, 81.6% vs. 85.1%, respectively; p=0.593 in RNA expression profile) (Table 1, Fig. 3A-C). Further survival analysis was also performed using the combination of TP53 mutation and expression status. The 5Y DRFS rate in patients with missense mutation and low expression of TP53 was 50.0%, and that in patients with truncation/deletion mutations and high expression was 75.0%, lower than those in patients with other TP53 status (p=0.057) (Table 3, Fig. 3D).
Multivariate analysis was performed using stage and the combination of mutation and mRNA expression of TP53 (Supplementary Table 2). According to the results of this analysis, stage was still remained a possible prognostic indicator (p < 0.001). In terms of TP53 status, marginal statistical significance also remained in this analysis (p=0.057); in particular, patients with missense mutation and low TP53 expression showed a 6.7-fold increase in distant recurrence rate compared to patients with low TP53 expression without mutation (TP53 missense mutation and low mRNA expression: hazard ratio, 6.73; 95% confidence interval, 1.67 to 27.10; p=0.057, compared with wild type TP53 and high mRNA expression, respectively).
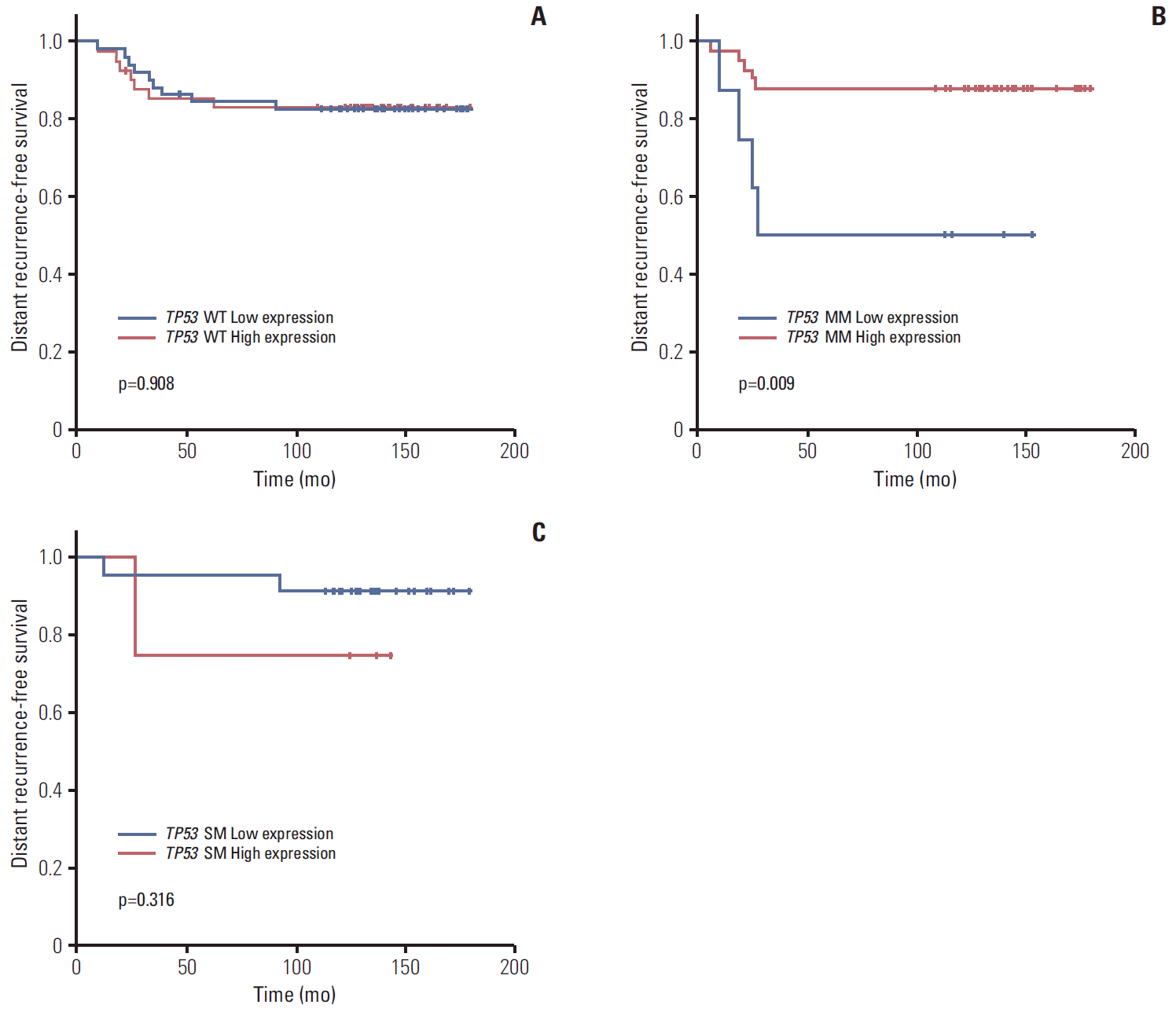
Subgroup analysis showed that expression level did not affect prognosis in patients with wild type TP53 (5Y DRFS: low vs. high, 81.8% vs. 83.3%,respectively; p=0.908) (Fig. 4A). However, in patients with missense TP53 mutations, low expression of TP53 was associated with poor prognosis (5Y DRFS: low vs. high, 50.0% vs. 87.8%, respectively; p=0.009) (Fig. 4B). In addition, high expression of truncation/deletion TP53 mutation indicated poor prognosis, despite a lack of statistical significance (5Y DRFS: low vs. high, 91.7% vs. 75.0%, respectively; p=0.316) (Fig. 4C).
Discussion
Mutations in TP53 gene are the most frequent genetic alterations in malignant epithelial tumors. Although many investigators have attempted to determine the significance of TP53 mutation and expression, there are no clear data regarding the significance of TP53 mutation in breast cancer. In our study, discrepancy between TP53 mutation and expression suggested worse prognosis in TNBC.
TP53 has been studied extensively in germline mutation in families affected with Li-Fraumeni syndrome [19] and in somatic mutation in sporadic breast cancer. Previous reports found that TP53 mutation loci were heterogeneous and that there were no actual hot spots [19,20]. Further studies have shown that TP53 mutations occur in specific exons out of 11 TP53 exons [21]. Recent large-scale genetic analyses demonstrated that the TP53 DNA-binding domain was the most frequently mutated area in the entire TP53 gene in breast cancer [13,22,23]. In these studies, the most common mutant loci encoded R273C, R175H, Y220C, and R248Q amino acid changes in breast cancer patients. A recent study demonstrated that only missense mutations in the DNA-binding domain affected the outcome of breast cancer patients [16]; however, these studies were conducted using all breast cancer subtypes. Our study also found that most mutations occurred in the TP53 DNA-binding domain (73 of 77 cases) and that sequences encoding Y200C, R248Q, and R273C amino acid loci were hot spots for TP53 mutations. However, we found no relationship between mutations in the DNA-binding domain and prognosis of TNBC patients.
We also found a strong association between mRNA and protein expression, and mutation status of TP53, in contrast with previous studies reporting no relation between TP53 mutation and expression [16,24]. However, previous studies were conducted without consideration of breast cancer subtype, and did not categorize TP53 mutations with respect to expected functional status after base substitution. In this study, p53 expression showed strong correlation with mutation status. In addition, TP53 mutations expected to result in functional loss showed lower expression, and missense mutations showed higher expression than wild type TP53. We also analyzed p53 expression level in the group with missense mutations in regard to mutation loci. In this analysis, mutation loci did not affect p53 expression status whether mutation loci were the R175H or R273H amino acid changes, or not [9]. Accordingly, our classification of TP53 mutation might be reasonable, and could predict expression status.
We also found that some TNBC patients lacked p53 expression despite having wild type TP53, which could be explained by a potential technical error of IHC. However, post-translational modification is another reasonable factor to result in a loss of p53 expression without mutation. Although epigenetic methylation sequencing analysis was not performed in our study, transcriptional and epigenetic modifications of TP53 are known mechanisms that could cause deregulation of TP53 [25,26]. These previous studies showed that transcriptional regulation of TP53 suppressed gene expression in both tumor samples and transformed cell lines without TP53 mutation. Accordingly, this discrepancy in our study also has a plausible explanation using post-translational modification of p53.
In this study, we examined the connection between TP53 status and patient prognosis. Even though TP53 status could not predict individual patient prognosis, TNBC patients with discrepancy between expression and mutation status of TP53, particularly missense mutation with low expression level, were found to have poor prognosis. We might suppose that low expression in missense TP53 mutations was caused not by mutation, but by epigenetic alteration of TP53, and that epigenetic alteration could affect patient prognosis. However, we were unable to explore the post-transcriptional status of TP53, which is a limitation of our study.
We could not perform subcategorization according to biological characteristics, which might be another limitation of our study. Because TNBC is a heterogeneous group of diseases with distinct biological characteristics [27], subcategorization of TNBC could be helpful in prediction of TNBC prognosis. Indeed, we tried to classify TNBC using gene expression by heterogeneous biology [28].
This study is the first study to classify TP53 mutation status according to categories defined by predicted functional activity, and the first report to show that discrepancy between p53 expression and mutation status of TP53 may be a potential prognostic marker in TNBC patients. Therefore, both DNA sequencing and evaluation of mRNA expression may be necessary to predict prognosis of TNBC patients. Further epigenetic study will be needed to clarify the mechanisms underlying discrepancy between expression and mutation status.