INTRODUCTION
Raf was found to be a cellular oncogene that is transduced by transforming retroviruses (1). The Raf-1/mitogen-activated protein (MAP) kinase pathway is now considered to be a major route for the transmission of signals from Ras to the transcription factor in the nucleus (2). It has been well-established that in addition to the association of Raf with GTP-charged Ha-Ras, additional phosphorylations on tyrosine and serine/threonine residues are required for the activation of Raf-1 kinase (3). However, the detailed molecular mechanism(s) involved in the cellular regulation of Raf-1 remains to be fully elucidated. Among a number of factors that mediate the activation of Raf-1 kinase (4), protein kinase C (PKC) has been demonstrated to regulate the Ha-Ras-MAP kinase pathway in a variety of systems ranging from yeast to higher eukaryotes (5). PKC has been extensively studied as a second messenger transducing diverse signal that's involved in cell proliferation and the activation of cellular functions, differentiation and even apoptosis (6).
The PKC family is composed of at least 11 members that are classified into three major groups: conventional PKC (cPKC) that includes PKC-α, -β1, -β2 and -γ; novel PKC (nPKC) that includes PKC-δ, -ε, -ι, -θ and -µ; and atypical PKC (aPKC), that includes PKC-ζ and -λ (7). It is believed that the cPKC class is important for 'short-term' receptor-mediated cellular responses to transient changes in cytosolic calcium, e.g., hormone secretion, and neuromediator release, while the nPKC class is likely to be involved in 'long-term' cellular regulation, e.g., cell growth, differentiation and tumor promotion (8). In mammalian cells, cPKC-α and nPKC-ε have been shown to activate Raf-1 (3). It has been shown that PKC-ε is likely to exert its transforming action by interacting with the ras signaling pathway at the level of ras-Raf-1 interaction (3,9). Interestingly, the closely related PKC-δ isoform displays cellular effects that are far different from those shown by PKC-ε. There is much data to support the hypothesis that PKC-δ acts as a negative regulator of cell proliferation and transformation (10).
Here we have employed the pharmacological PKC activator PMA to determine the possible involvement of PKC activation in the modulation of Raf-1 kinase activity. PMA acts as a substitute for diacylglycerol (11) and it directly activates PKC both in vitro and in vivo (12). The results presented here suggest that two different stimulatory signals derived from PMA and PDGF can converge at PKC to activate Raf-1 kinase, but they induce contrasting effects on cell proliferation.
MATERIALS AND METHODS
1) Materials
The rabbit polyclonal anti-Raf (C-12) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA), while the anti-phospho-PKC (pan) was obtained from Cell Signaling Technology (Denvers, MA). The protein A-agarose was from Boehringer Mannheim. Dulbecco's modified Eagle's medium (DMEM), fetal calf serum (FBS), penicillin and streptomycin were purchased from GIBCO-Invitrogen (Carlsbad, CA). The reagents for SDS-polyacrylamide gel electrophoresis were from Bio-Rad (Hercules, CA). [γ-32P]ATP (3,000 Ci/mmol) was purchased from New England Nuclear (Boston, MA). N-acetyl-L-cysteine and wortmannin were purchased from Calbiochem (San Diego, CA). The PDGF was from R&D systems Inc (Minneapolis, MN). The PMA was obtained from Sigma (St. Louis, MO). The PMA was dissolved in DMSO and it was freshly diluted for each experiment.
2) Mammalian Cell Culture and transient transfection
The parental and v-Ha-ras transfected NIH 3T3 cells were maintained at 37℃ in DMEM supplemented with 10% FBS, penicillin/streptomycin and glutamine. Where indicated, the cells were transiently transfected with the pMTH vector that encodes dominant-negative (DN) PKC-ε with using Lipofectamine 2000 (Invitrogen) in Opti-minimal essential medium I (Invitrogen) according to the manufacturer's protocol. After 24h, the transfected cells were serum-deprived overnight prior to treatment with PMA.
3) Cell growth assay
The cell proliferation reagent WST-1 was used for the quantitative determination of cellular proliferation (Roche Molecular Biochemicals, Germany). For the proliferation assays, the cells were plated in quadruplicate into 96-well microliter plates at 5×103 cells/well and then they were treated with PMA at 37℃ in a humidified 5% CO2/95% air incubator. After incubation for 2 or 3 days, the cells were incubated for an additional 4 h in the presence of WST-1 labeling mixture (10 µl per well). The absorbance of the samples, as compared with using a background control (medium alone) as a blank, was measured at 450 nm by using an ELISA reader (Molecular Devices, Sunnyvale. CA).
4) Preparation of Cell Lysates
The whole cell lysates were prepared as follows. The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and they were harvested by resuspending the cell pellet in lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA containing 1 mM phenylmethylsulfonyl fluoride, 20 µg/ml aprotinin, 10 µg/ml leupeptin, 20 mM β-glycerophosphate and 2 mM sodium fluoride). The cell lysates were clarified by centrifugation at 15,000×g for 10 minutes at 4℃, and their protein concentrations were determined with the aid of a BCA protein assay reagent kit (Pierce; Rockford, IL).
5) Immunoprecipitation and in vitro Raf-1 kinase activity assay
The Raf-1 proteins were specifically immunoprecipitated from the whole cell lysates; they were washed three times with lysis buffer and once with kinase buffer (20 mM Tris, pH 7.4, 20 mM NaCl, 1 mM dithiothreitol, 10 mM MgCl2). The Raf-1 kinase activity was measured by phosphorylation of the recombinant MEK (Santa Cruz Biotechnology, Santa Cruz, CA) as previously described (13). The washed immunoprecipitates were incubated in 40 µl of kinase buffer containing 10 mM ATP, 1 µg of the recombinant MEK and 5 µCi of [γ-32P] ATP at 30℃ for 30 min in the presence of the MEK inhibitor PD 98059, which was used to inhibit the autokinase activity of MEK-1. The assays were terminated by the addition of gel-loading buffer. The samples were resolved by 7.5% SDS-PAGE, and the phosphorylated MEK protein bands were visualized by autoradiography. The bands were quantified using Quantity One analysis software, Version 4.4 (Bio-Rad).
RESULTS
1) The involvement of PKC in Raf-1 kinase activation
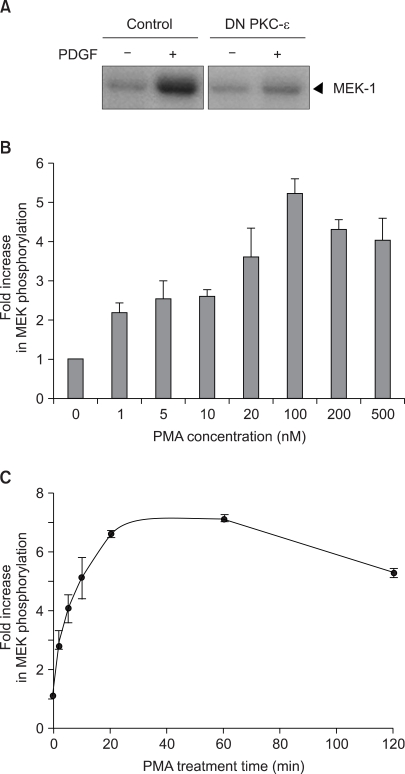
The activation of PKC has been shown to play an important role in the growth factor activation of Raf-1 kinase (3). Consistent with this previously report, the expression of the kinase-defective, DN mutant of PKC-ε K436R led to a significant inhibition of PDGF-induced Raf-1 kinase activation (Fig. 1A). To further confirm the activation of Raf-1 kinase by PKC, the impact of the pharmacological PKC activator PMA on Raf-1 kinase was investigated in v-Ha-ras transformed NIH 3T3 cells, which were found to be more susceptible to PMA activation than its parental NIH 3T3 cells. PMA was found to activate Raf-1 kinase with the induction first becoming apparent at 1 nM PMA in the v-Ha-ras transformed cells. The maximum stimulation of Raf-1 kinase was observed at the concentration of 100 nM PMA (Fig. 1B). The kinetics of Raf-1 activation following exposure of the v-Ha-ras transformed cells to PMA are shown in Fig. 1C. Promotion of the PMA-induced activation of Raf-1 was maximal with 20 min of PMA treatment, and this level of activation was sustained during at least 2 h of exposure to PMA.
2) The inhibitory effect of PMA on cell growth
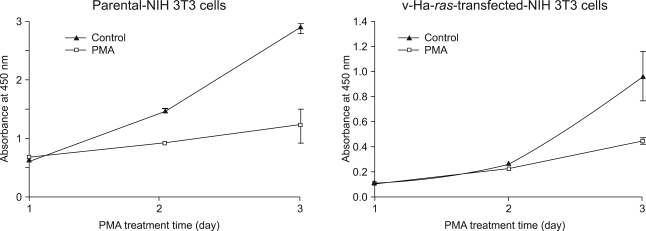
Earlier studies have shown that the increase in Raf-1 kinase activity is a significant event in PDGF-induced mitogenesis (14). Thus, to evaluate for the ability of PMA to alter cell proliferation via PKC activation, the parental cells and its v-Ha-ras transfected NIH 3T3 cells were grown on a 96-well plate and the numbers of viable cells were quantified using a colorimetric assay. Contrary to the fact that PDGF induces cellular DNA synthesis in NIH 3T3 cells (15), PMA produced a time-dependent inhibition of proliferation in both cell lines. (Fig. 2) The suppression of parental NIH 3T3 cell proliferation was first observed on day 2. However, treating the v-Ha-ras-transformed cells with PMA resulted in delayed inhibition of cell growth. Of added interest was the finding that compared to the results obtained with the parental NIH 3T3 cells whose growth was completely arrested, the v-Ha-ras-transformed cells continued to grow, but at a slower rate than the untreated cells.
3) The effect of PI3 kinase inhibitor wortmannin on PMA-induced Raf-1 kinase activation
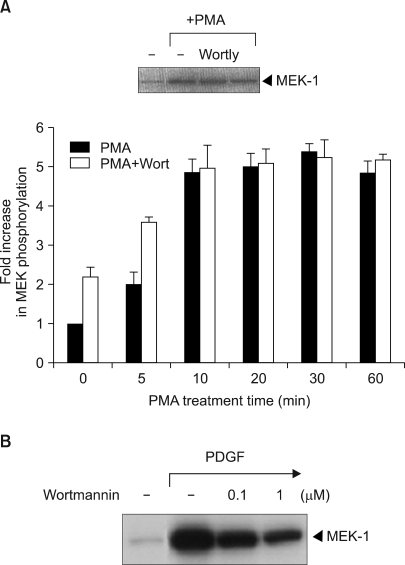
We next investigated if phosphatidylinositol-3-kinase (PI3 kinase), which is an upstream activator of Raf-1, is involved in PMA-induced Raf-1 kinase activation. Wortmannin is a fungal metabolite that irreversibly modifies the catalytic domain of PI 3-kinase and it renders the enzyme catalytically inactive (16). It was shown that wortmannin did not significantly block the PMA-induced activation of Raf-1 (Fig. 3A). Also, with using LY294002 (2-(4-morphlinyl)-8-phenyl-4H-1-benzopyran-4-one), which is another chemically different inhibitor of PI3K, it was suggested that PI 3-kinase was not involved in the PMA-induced Raf-1 kinase signaling pathway (Fig. 3A, inset). These results established that the PMA-induced activation of Raf-1 was PI3 kinase-independent. Conversely, wortmannin effectively blocked the PDGF-induced Raf-1 kinase activation (Fig. 3B).
4) The role of reactive oxygen species in PMA-induced Raf-1 kinase activation
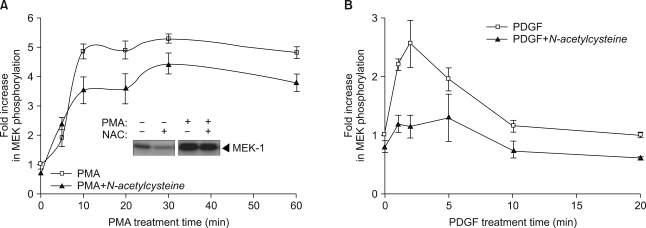
Since ROS has been reported to induce prolonged activation of PKC (17), we next investigated the effect of the chemical antioxidant N-acetylcysteine on PMA-mediated Raf-1 kinase activation. Pretreatment with N-acetylcysteine had little inhibitory effect on PMA-induced Raf-1 kinase activation (Fig. 4A), suggesting that ROS may not be required for the signaling pathways leading to Raf-1 activation by PMA. Conversely, PDGF-induced Raf-1 kinase activation was completely inhibited by pretreatment with N-acetylcysteine (Fig. 4B).
DISCUSSION
PKC is known to play important roles in such physiological events as cell differentiation and proliferation (18). In this study, exposure of the v-Ha-ras-transformed NIH 3T3 cells to a pharmacological PKC activator PMA was found to result in the stimulation of Raf-1 kinase. The mouse fibroblast NIH 3T3 cells employed in our studies express the phorbol ester-sensitive isotypes cPKC-α, nPKC-ε and nPKC-δ (18). Among these PKC isotypes, we are especially interested in nPKC-ε, which is the only isoform that has its oncogenic potential mediated through its interaction with Raf-1 kinase. Our previous reports showed that the PMA-induced activation of Raf-1 kinase was suppressed in cells that expressed the kinase deficient, DN mutants of PKC-ε-K436R (13). This present study also demonstrated that the expression of the kinase-defective, DN mutant of PKC-ε K436R inhibited the PDGF-induced Raf-1 kinase activation (Fig. 1A), supporting the hypothesis that PKC-ε is actually involved in the physiological Raf-1 kinase signaling pathway. Our findings are in line with the work of Perletti et al., who found a marked increase of Raf-1 phosphorylation in PKC-ε-transformed colon epithelial cells (9). Some researchers have also determined the in vivo functional specificity of the PKC-ε in PMA-activated PKC signals (19). On the other hand, Schonwasser et al. (20) presented data favoring indirect activation rather than direct activation of Raf-1 kinase by PKC. It was also reported that PKC-ε induces the secretion of growth factors, which then activate Raf-1 via an autocrine loop (21). In addition to these mechanism(s), PKC was recently found to switch the Raf kinase inhibitor from Raf-1 to G-protein-coupled receptor kinase 2 (GRK-2), which resulted in Raf-1 kinase activation (5).
Despite this discovery of the involvement of PKC-ε in Raf-1 kinase activation, the molecular mechanism by which PMA-induced PKC activation transmits the signal to Raf-1 kinase remains unclear. Moriya et al. (22) showed that the effect of PDGF that resulted in translocation of PKC-ε could be attributed to the ability of PDGF to activate PI 3-kinase. In fact, wortmannin, which is PI 3-kinase inhibitor, inhibited Raf-1 kinase activation by PDGF, suggesting that PI3 kinase is involved in the signaling pathway that mediates PDGF activation of Raf-1. Thus, the impact of wortmannin for PMA signaling was investigated in order to demonstrate that PI 3-kinase is actually involved in the PMA-induced Raf-1 kinase signaling pathway. Rather unexpectedly, wortmannin showed no effect on the Raf-1 kinase activation by PMA. Consistent with this result, other researchers (23) have shown that activation of PI 3-kinase could be dissociated from the Raf-1 downstream signaling pathway, although a few studies have linked the activation of PI 3-kinase with the activation of PKC isoforms in whole cells (22).
ROS has been reported to induce prolonged activation of PKC (17). Many cell types produce ROS in response to a variety of growth factors such as PDGF (24) and epidermal growth factor (EGF) (25). As expected, PDGF-induced Raf-1 kinase activation was completely blocked by pretreatment with N-acetylcysteine. Conversely, no apparent inhibition of Raf-1 kinase activation by PMA was observed with using N-acetylcysteine. Taken together with the result from the wortmannin experiments, these observations provide evidence that the regulation of the Raf-1 cascade by both PDGF and PMA might be intimately linked and they converge at PKC-ε through different upstream pathways. However, PDGF induced the mitogenic signaling through the PKC-ε activation, whereas PMA caused growth arrest.
CONCLUSION
The results presented here suggest that two different stimulatory signals derived from PMA and PDGF can converge at PKC to activate Raf-1 kinase, but these signals induce the contrasting effects on cell proliferation. The signaling mechanism underlying this effect needs to be further identified.