Introduction
Histone deacetylase (HDAC) deacetylases lysine residues on both core histones and non-histone targets. So far, 18 genes were found to encode HDAC family, which are grouped according to class I to IV [1]. Aberrant regulation of classic HDACs (class I, II, and IV) has been reported in various cancer types suggesting significance of HDAC in carcinogenesis. Thus, classical HDACs have been a new target for anti-cancer drug development. However, pan-HDAC inhibitors lack isotype selectivity, simultaneously inhibiting multiple HDAC isotypes. Unselective inhibition of multiple HDAC isotypes might underlie numerous adverse effects demonstrated in clinical studies [1].
Radiotherapy is an integral component in an armamentarium of cancer treatments, and identification of effective radiosensitizers has been an area of intense research. However, development of radiosensitizers has been hampered by their unsatisfactory efficacy revealed in clinical trials. In the last two decades, various HDAC inhibitors of diverse structural backbones have been reported to enhance radiation lethality in cancer cells [2-10]. These observations suggest that any molecules with HDAC inhibitory activity induce radiosensitization. Thus, HDAC might provide a useful target for modulation of cellular radiosensitivity, implying that HDAC inhibitors have a therapeutic potential to overcome radiation resistance of cancers. Relation of class selectivity of HDAC inhibitors to their radiosensitizing properties has already been demonstrated [11]. Although pan-inhibitory profile of HDAC inhibitors might be preferred for therapeutic application, the lack of isotype-specificity hampers investigation into mechanisms underlying radiosensitization by HDAC inhibition. Suppression of DNA double strand break (DSB) repair has been suggested as a mechanism underlying radiosensitization by HDAC inhibitors [4,5,7,10]. However, the mechanisms regarding involvement of different HDAC isotypes in regulation of DNA DSB repair pathways are not fully elucidated.
The aim of the current study was to identify HDAC isotypes responsible for radiosensitization induced by HDAC inhibition. RNA interference was applied for selective inhibition of 11 members of classical HDACs (HDAC1 to HDAC11). This allowed us to determine the relative contributions of each HDAC isotype towards cellular response to ionizing radiation. Isotype-specific inhibition using specific siRNAs targeting HDAC isotypes showed significant attenuation of protein expression of corresponding HDAC. Specific inhibition of class I HDAC (HDAC1 and HDAC3), class II HDAC (HDAC4, HDAC6, HDAC 7, and HDAC10), or class IV HDAC (HCAC11) decreased clonogenic survival of SQ20B cells following ionizing radiation. Inhibition of these isotypes resulted in downregulation of various histonedependent or non-histone target proteins implicated in radiation response. Correlation was observed between radiosensitization by HDAC inhibition and interference of DNA DSB repair. Inhibition of some HDAC isotypes resulted in downregulation of key molecules involved in DSB repair and prosurvival pathway.
Materials and Methods
1. Cell culture
The SQ20B cell line was obtained from a patient with recurrent laryngeal carcinoma as described previously [12]. Cells were maintained and cultured using standard techniques at 37°C in 5% (v/v) CO2 using culture medium recommended by the supplier. In all experiments, different cell populations were first cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum.
2. Synthetic small interfering RNAs
Synthesis and analysis of target sequence-specific siRNAs were performed as previously reported [12]. Specific siRNAs against each HDAC isotype and nonspecific control siRNA were purchased from a commercial vendor (Bioneer, Daejeon, Korea). The target sequences for the siRNAs used in this study were as follows: control, ACG AAA UUG GUG GCG UAG G (dTdT); HDAC1, UCA UCC UCU GUU UUG ACU C (dTdT); HDAC2, ACU GAG CUC AGU UUC CGU C (dTdT); HDAC3, UAG AGG GAU AUU GAA GCU C (dTdT); HDAC4, AAG UCU GAG CCU CGA UCA G (dTdT); HDAC5, AGA CGA UAA CAG ACA GAC G (dTdT); HDAC6, UCU AGG CUG UGA ACC AAC A (dTdT); HDAC7, AUC AGU UGC UGC GUC AUG U (dTdT); HDAC8, AAG GAU GUA CUU AAG ACA C (dTdT); HDAC9, UUU GCU GUC GCA UUU GUU C (dTdT); HDAC10, UGA UAU CCG GCG UUG UCA G (dTdT); and HDAC11, UGU CGU CCA UGA AGU CUC G (dTdT).
3. Transfection of siRNA and clonogenic survival assay
Transfection of siRNA into SQ20B cells was performed using a previously reported method [12]. Briefly, 1×105 to 2×105 cells were plated into 6-well culture plates. When cells were 30%-40% confluent, the medium was changed to antibiotic-free medium. Thirty to 60 μL of each annealed oligo duplex (25 mmol/L concentration) in 235 μL of reduced serum medium (Opti-MEM, Life Technologies, Stratagene, La Jolla, CA) was transfected into cells using 15 to 30 μL of Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Transfection efficiency, monitored by fluorescence microscopy and fluorescence-activated cell sorting analysis for intracellular uptake of Cy-5–labeled oligonucleotides at the initial phase of this study was consistently over 80%.
The method used for clonogenic assay was previously reported [8]. Two to 4 days after transfection, cells were trypsinized, and a specified number of cells were seeded in six-well culture plates for irradiation. Cells were exposed to graded doses of radiation using a medical linear accelerator (Varian Medical Systems, Palo Alto, CA) at a dose rate of 2.46 Gy/min. Colonies were fixed with methanol, stained with 0.5% crystal violet, and counted 14 to 21 days after irradiation. For generation of survival curves, surviving fraction data was fitted to a linear-quadratic model using the Kaleidagraph software (ver. 3.51, Synergy Software, Reading, PA). Each point on the survival curve represents the mean surviving fraction from at least triplicate dishes. Sensitizer enhancement ratio (SER) was defined as the ratio of the isoeffective radiation doses with the transfection of siRNA against specific HDAC isotypes to doses with the transfection of nonspecific control siRNA at a surviving fraction of 0.5.
4. Immunoblot analysis
Cells were harvested using lysis buffer (iNtRON Biotechnology, Seoul, Korea). Proteins were solubilized by sonication and equal amounts of protein were separated on sodium dodecyl sulphate polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). Membranes were blocked in phosphate-buffered saline containing 0.1% Tween 20 and 5% powdered milk, and probed with the primary antibody against each HDAC isotype (HDAC1, HDAC3, HDAC4, HDAC5, HDAC6, and HDAC7 antibodies from Cell Signaling, Beverly, MA; HDAC8 and HDAC9 antibodies from Santa Cruz Biotechnology, Santa Cruz, CA; HDAC10 antibody from Sigma-Aldrich, St. Louis, MO; HDAC11 antibody from Abgent, San Diego, CA). Antibodies against Rad51, phospho-ataxia telangiectasia mutated (phospho-ATM), Aurora A, Aurora B, X-linked inhibitor of apoptosis (XIAP), and survivin were purchased from Cell Signaling Technology.
5. Immunocytochemistry
Immunocytochemistry was performed as described previously [9]. Slides were examined on an Axio Scope Imager A1 fluorescence microscope. Images were captured and acquired using AxioCam MR c5 and acquisition software AxioVision v.4.4 (Carl Zeiss, Gottingen, Germany).
Results
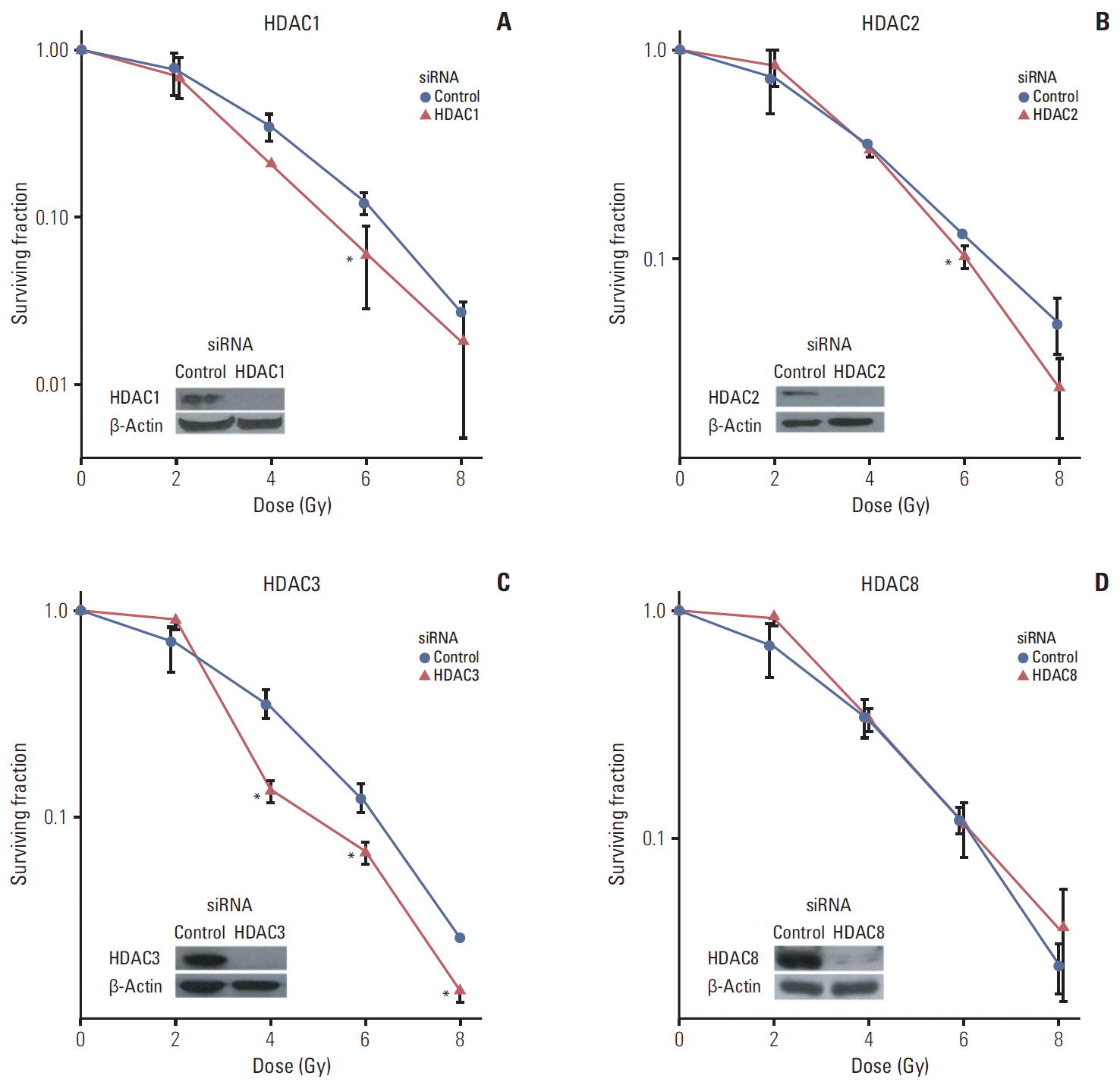
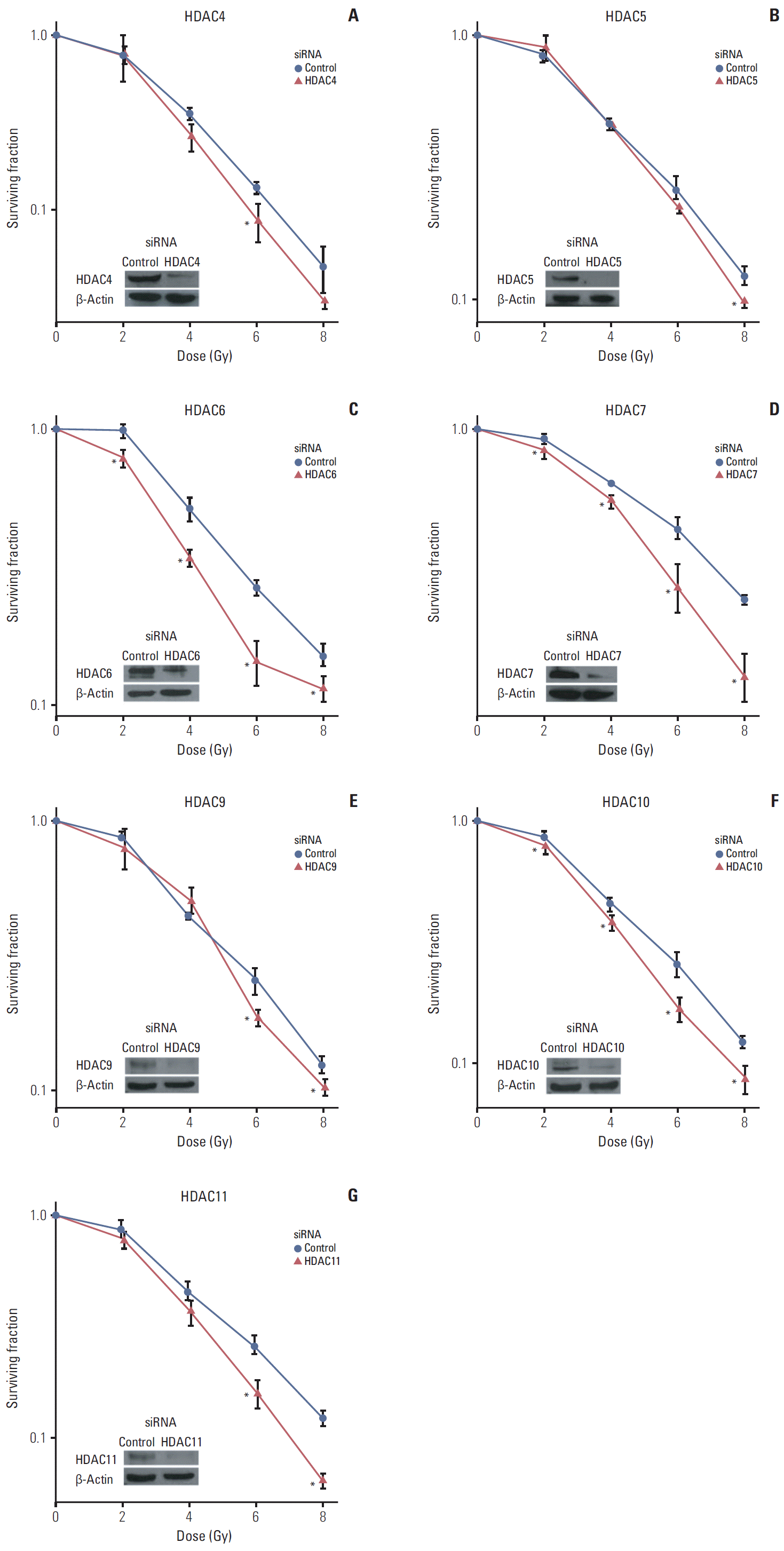
1. Selective inhibition of HDAC isotypes reduces tumor cell radiation survival
Transfection with siRNA against HDAC isotypes specifically inhibited expression of only corresponding isotypes (Supplementary Fig. S1). For direct evaluation of the differential contribution of HDAC isotypes to cellular radiation resistance, we examined the radiosensitivity of SQ20B cells treated with siRNA specific for various isotypes of class I, II, and IV HDAC. Our working criterion of radiosensitization was SER of 1.2 or greater. Of class I HDAC, selective inhibition of HDAC1 and HDAC3 resulted in decreased radiation survival in SQ20B cells to similar degrees (Fig. 1). Among members of class II and IV HDAC, specific inhibition of HDAC4, HDAC6, HDAC7, HDAC10, and HDAC11 resulted in reduced radiation survival (Fig. 2). SER from selective inhibition of these HDAC isotypes are summarized in Table 1.
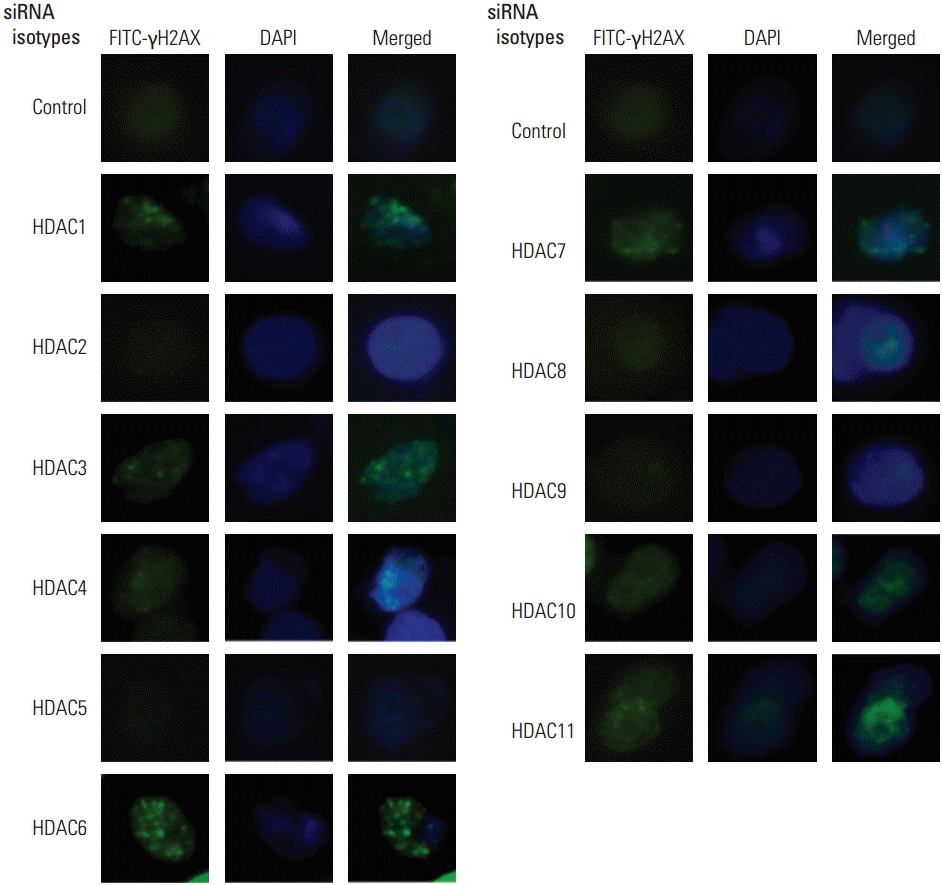
2. Selective inhibition of HDAC isotypes interferes with DNA repair proteins
As shown above, selective inhibition of a subset of HDAC isotypes can enhance radiation lethality in SQ20B cells. As an initial investigation into the mechanism responsible for this radiosensitization, we used immunoblot analysis of phosphorylated histone H2AX (γH2AX). Irradiation of SQ20B cells immediately induced γH2AX foci (data not shown). To determine the effect of HDAC isotype-specific inhibition on γH2AX, SQ20B cells transfected with siRNA against HDAC isotypes were irradiated to 8 Gy, and foci formation was evaluated 4 hours after irradiation. In cultures transfected with nonspecific control siRNA, the γH2AX level at 4 hours returned to unirradiated levels (Fig. 3, panels labeled as control). The elevated level of γH2AX foci formation remained constant in cells under selective inhibition of HDAC1, HDAC3, HDAC4, HDAC6, HDAC7, HDAC10, and HDAC11 (Fig. 3). The maintenance of γH2AX levels suggested that radiosensitization via selective HDAC isotype inhibition involves interference with DNA damage repair.
3. Isotype-specific inhibition of HDACs and various target proteins
As presented above, selective inhibition of some HDAC isotypes can augment radiation lethality in SQ20B cells. This suggests that interference in cellular radiation responses might underlie radiosensitization by siRNA targeting specific HDAC isotypes. Immunoblot analysis of proteins known to be associated with radiosensitivity was performed to identify targets affected by HDAC inhibition. Transfected SQ20B cells were irradiated to a single dose of 8 Gy. Proteins were extracted for Western blot 4 hours after irradiation. Rad51 is an early component of homologous recombination of DNA DSB repair. Isotype-specific inhibition of HDAC1, HDAC3, HDAC4, HDAC6, HDAC7, HDAC8, HDAC9, and HDAC10 resulted in downregulation of Rad51 in SQ20B cells (Fig. 4A). These findings implicate Rad51 as an intermediary of radiosensitization by HDAC inhibition. Next, we investigated the effect of selective HDAC inhibition on ATM phosphorylation. Selective suppression of HDAC1 resulted in down-regulation of phospho-ATM, while inhibition of HDAC2 had no apparent effect on its expression (Fig. 4B). HDAC inhibition was previously reported to induce degradation of Aurora kinases [13]. Selective inhibition of HDAC3 and HDAC4 resulted in downregulation of Aurora kinases B and A, respectively (Fig. 4C). Survivin and XIAP are members of inhibitors of apoptosis (IAP). Inhibition of survivin or XIAP was reported to result in radiosensitization of lung cancer cells [14]. Inhibition of HDAC5, however, had no influence on either Aurora A or Aurora B expression (Fig. 4C). We found that selective inhibition of HDAC7 resulted in attenuated expression of both survivin and XIAP (Fig. 4D).
Discussion
We reported on the first comprehensive investigation into the contribution of HDAC isotypes towards radiosensitization by HDAC inhibition. Using isotype-specific siRNA, we found that only a subset of HDAC isotypes are involved in modulation of radiosensitivity of human laryngeal cancer cells. Radiosensitization by isotype-selective HDAC inhibition was coupled with delayed repair of DNA DSB. In addition, selective inhibition of some isotypes resulted in downregulation of key proteins involved in cellular radiation responses and DNA damage repair.
Classic HDACs are a promising category for anti-cancer drug targets. Apart from their anti-cancer effects, reports that various HDAC inhibitors enhance radiation lethality of cancer cells in both animal and in vitro models have been consistent [2-10]. Given the observations using inhibitors of varied structural backbones, it is assumed that HDAC inhibition generally induces radiosensitization. However, the mechanisms of HDAC isotypes regulating cellular radiosensitization are not fully understood. We previously reported the class difference of HDAC inhibitors in sensitizing tumor cells to ionizing radiation. Trichostatin A, which inhibits both class I and II of HDAC, was a more potent sensitizer than SK-7041, a class I HDAC inhibitor. Splitomicin, an inhibitor of class III HDAC, had no apparent effect on cellular radiosensitivity [11]. However, relative contribution of HDAC isotypes has not been addressed comprehensively. Most pharmacological HDAC inhibitors lack isotype-selectivity, and inhibit a wide range of HDAC isotypes to varying degrees [1]. Thus, most reports using HDAC inhibitors are inherently inadequate to interrogate relationships of specific HDAC isotypes with radiosensitivity.
Instead of HDAC inhibitors, specific siRNA was used against a panel of HDAC isotypes. In SQ20B cells transfected with isotype-selective siRNA, inhibition of HDAC1, HDAC3, HDAC4, HDAC6, HDAC7, HDAC8, HDAC10, and HDAC11 resulted in increased radiation lethality (Figs. 1 and 2). Suppression of the remaining HDAC isotypes had no apparent effect on cellular radiosensitivity. Current observations suggest that members of the HDAC family may unevenly contribute to radiosensitization by HDAC inhibition. Other investigators have implicated a specific HDAC isotype in cellular radiation responses. Silencing of HDAC4 via RNA interference was reported to result in radiosensitization of HeLa cells [15]. HDAC4 silencing decreased expression of 53BP1 and abrogated radiation-induced G2-phase delay. Geng et al. [4] reported translocation of HDAC4 from the cytoplasm into the nucleus of lung cancer cells following irradiation. Treatment with LBH589, an HDAC inhibitor, increased cellular radiosensitivity and blocked nuclear translocation of HDAC4. These results fit with our observation that selective HDAC4 inhibition enhanced radiation lethality in SQ20B cells. Contrary to our previous report [11], we found that inhibition of some class I (HDAC2 and HDAC8) and class II (HDAC5 and HDAC9) had little influence on radiosensitivity. Thus, it is plausible that HDAC inhibition might induce radiosensitization in an isotype-specific, not a class-dependent manner. However, isotype-specificity determining HDAC-mediated sensitization is poorly understood.
Irradiation arrests cell cycle progression at G2/M phases, and induces γH2AX foci in the nucleus. γH2AX foci are formed at DNA DSB, and their temporal dynamics serve as an indicator of the DNA repair process. HDAC inhibition has been consistently reported to abrogate radiation-induced cell cycle arrest at the G2/M phase [16] and delay clearance of radiation-induced γH2AX foci [4,5,7,9]. We observed that radiosensitization by selective inhibition of several HDAC isotypes was accompanied by impediment of delayed disposal of radiation-induced γH2AX foci in SQ20B cells. However, inhibition of other class I (HDAC2 and HDAC8) and class II (HDAC5 and HDAC9) HDAC isotypes apparently neither increased radiosensitivity nor affected clearance of γH2AX foci (Fig. 3). Of note is that siRNA against these isotypes showed no apparent influence on clearance of radiation-induced γH2AX foci (Fig. 3). Taken together, these might suggest that interference with DNA DSB repair is an integral part of HDAC inhibition-induced radiosensitization. Our observations showed that unhindered activity of certain HDAC isotypes is essential for full functionality of cellular DNA damage repair machinery.
DNA DSB are major lethal lesions caused by ionizing irradiation, and two basic pathways are responsible for DSB repair in eukaryotic cells: homologous recombination and nonhomologous end joining [17]. Rad51 is recruited to DSB sites via interaction with BRCA2, and plays a central role in initiation of homologous recombination. We found that inhibition of a subset of HDAC isotypes resulted in diminished expression of Rad51 following irradiation in SQ20B cells. Apparent downregulation of Rad51 followed transfection of cells with siRNA against all HDAC isotypes except HDAC2, HDAC5, and HDAC11 (Fig. 4A). Our observations imply that selective inhibition of some HDAC isotypes impedes the homologous recombination pathway by downregulation of Rad51. However, siRNA against HDAC11 enhanced radiation lethality without apparent influence on Rad51 expression (Figs. 2G and 4A). Thus, it is unlikely that downregulation of Rad51 and resultant interference with homologous recombination is the sole mechanism underling sensitization by HDAC inhibition.
ATM belongs to a family of kinases with sequence homology to phosphoinositide 3-kinase. Irradiation leads to activation of ATM via its autophosphorylation and dissociation of dimeric forms [17]. We observed that selective inhibition of HDAC1 decreased phospho-ATM in irradiated SQ20B cells, while HDAC2 inhibition resulted in no apparent change in ATM phosphorylation (Fig. 4B). Results of clonogenic survival experiments and immunoblotting analyses suggest that inhibition of HDAC1 induces radiosensitization by suppression of ATM autophosphorylation following irradiation. ATM interacts with HDAC1 both in vitro and in vivo. Irradiation strengthens an association between ATM and HDAC1, and HDAC inhibitor disrupts the interaction between two molecules [18]. Thus, our findings show that interaction of HDAC1 with ATM is required for autophosphorylation of the latter following irradiation. Since ATM is a regulator of both homologous recombination and nonhomologous end joining pathways, HDAC1 inhibition likely impedes repair of DNA DSB and results in radiosensitization. Rad51 expression and ATM phosphorylation was not affected by selective inhibition of HDAC2 in irradiated SQ20B cells (Fig. 4A and B). Thus, it may be conjectured that HDAC2 inhibition is irrelevant with diminution of DNA repair pathways. This postulation is corroborated by our observation that selective HDAC2 inhibition resulted in no radiosensitization (Fig. 1B).
Aurora kinases are serine/threonine kinases known as key mitotic regulators in maintenance of genomic stability, and aberrant regulation of Aurora A and Aurora B kinases has been reported in various human cancers [19]. Inhibition of Aurora kinases has been shown to enhance radiosensitivity in cancer cells [20,21]. We observed that selective inhibition of HDAC3 and HDAC4 resulted in downregulation of Aurora B and Aurora A kinases in irradiated SQ20B cells (Fig. 4C). Since inhibition of both HDAC3 and HDAC4 increased cellular susceptibility to radiation lethality (Figs. 1C and 2A), downregulation of Aurora kinases might underlie radiosensitization by inhibition of these HDAC isotypes. Our observation agrees well with that of Cha et al. [13] using LBH589, an HDAC inhibitor, who showed that treatment with LHB589 resulted in degradation of both Aurora A and Aurora B by targeting HDAC3 and HDAC6. Taken together, these findings suggest that downregulation of Aurora kinases might contribute to radiosensitization by selective inhibition of HDAC3, HDAC4, and HDAC6.
Both survivin and XIAP, members of IAP, play a major role in protecting cells from apoptotic signals. Thus, both survivin and XIAP are considered to confer marked resistance to cancer treatment. Survivin expression is a marker of tumor resistance to radiotherapy. High expression of survivin was associated with higher risk of local recurrence in rectal cancer patients after preoperative chemoradiotherapy [22]. Accordingly, inhibition of IAP has been investigated as a strategy to overcome tumor resistance to cancer treatment. Enhanced radiation lethality in human cancer cells by knockdown of survivin and XIAP using antisense oligonucleotides has been well demonstrated [14,23,24]. In a lung cancer xenograft model, combined inhibition of both survivin and XIAP resulted in increased delay of tumor growth after tumor irradiation [24]. However, the role of IAP as a mediator of HDAC-related radiosensitization has not been reported. We observed that selective inhibition of HDAC7 resulted in downregulation of both survivin and XIAP in SQ20B cells (Fig. 4D). These findings imply involvement of suppression of IAP in radiosensitization by HDAC inhibition.
The current study is the first comprehensive investigation into radiosensitization by isotype-specific HDAC inhibition. Our findings showed that various HDAC isotypes are differentially implicated in determining cellular radiosensitivity. Interference with DNA DSB repair is likely the pivotal mechanism underlying radiosensitization by isotype-selective HDAC inhibition. It is likely that HDAC inhibition impedes homologous recombination by downregulation of Rad51, although nonhomologous end joining may also be affected. However, disruption of the DNA repair pathways is likely not the sole mechanism of modulation of radiosensitivity by HDAC inhibition. To the contrary, radiosensitization by HDAC inhibition may be mediated by downregulation of various pro-survival, mitotic regulatory proteins. Given both the isotype diversity and wide range of biological targets of HDAC, it is postulated that HDAC inhibition affects multiple cellular pathways and perturbs radiation responses, which converges onto hindrance of DNA damage repair.
These results expand the growing evidence of the differential roles of individual HDAC isotypes, and provide proof of the concept of optimal targets for effective radiosensitization that combine a preferred functional profile with least unwanted side effects.
Conclusion
HDAC isotypes are differentially implicated in determining cellular radiosensitivity. Only a subset of classic HDAC isotypes are involved in regulating cellular response to ionizing radiation via interaction with proteins of DNA DSB repair and pro-survival pathways. The current study provides a rationale for the need of optimized approach for future development of HDAC-mediated radiosensitization strategies as well as an insight into the differential role of HDAC isotypes in modulating cellular radiosensitivity.