Introduction
von Hippel–Lindau (VHL) disease is an autosomal dominant inherited tumor syndrome associated with mutations of the VHL tumor suppressor gene located on chromosome 3p25 [1,2]. The loss of functional VHL protein contributes to tumorigenesis [3]. This condition is characterized by development of benign and malignant tumors in the central nervous system (CNS) and the internal organs, including kidney, adrenal gland, and pancreas [1]. We herein describe the case of a 74-year-old man carrying the VHL gene mutation who was affected by simultaneous colorectal adenocarcinoma, renal clear cell carcinoma, and hemangioblastomas in CNS.
Case Report
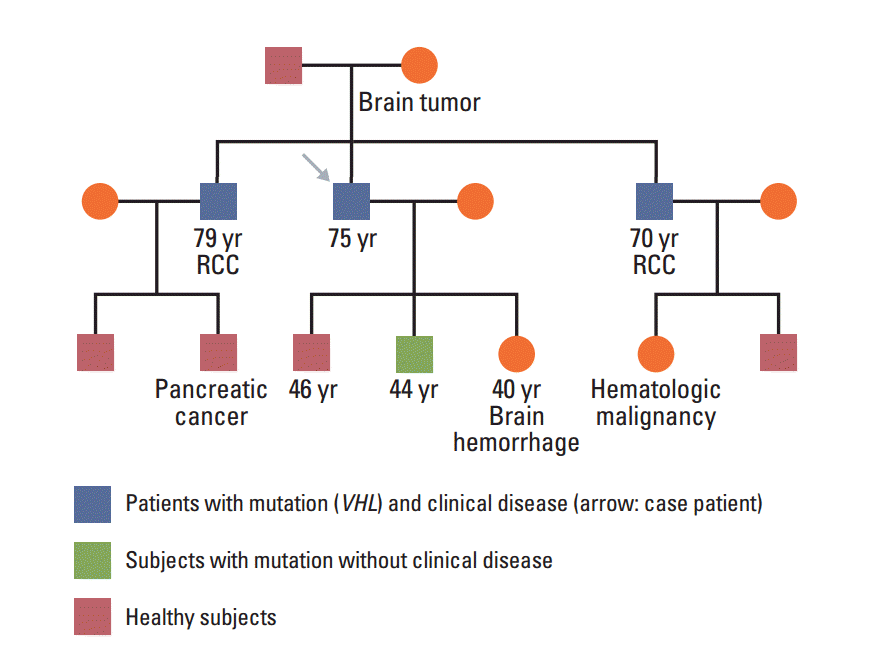
In January 2013, a 74-year-old man was admitted to the Department of Neurosurgery at Yonsei University College of Medicine due to gait disturbance and tingling sensation of both legs. The patient was receiving beta-adrenergic antagonist and calcium channel blocker for arterial hypertension. He had lost his vision in the left eye due to cataract and amblyopia in the right eye due to retinitis which had occurred 30 years ago. The patient’s family history included a mother with a brain tumor of unconfirmed pathology, and both brothers with renal cell carcinoma with VHL gene mutation (Fig. 1). Serum carcinoembryonic antigen (CEA) was elevated to 11.78 ng/mL (normal value, 0.0 to 5.0 ng/mL). Although he had no history of diabetes mellitus, serum glucose level was 462 mg/dL and HbA1c was 11.7%, which led to a new diagnosis of diabetes mellitus, and initiation of insulin therapy. Whole spine magnetic resonance imaging (MRI) showed multiple enhancing nodular lesions of the spinal canal suspected leptomeningeal metastasis of unknown primary malignancy (Fig. 2A). Positron emission tomography whole body scan showed intense fludeoxyglucose uptake in a 3-cm-sized heterogeneous mass in the right kidney and focal mural thickening in the sigmoid colon (Fig. 2B and C). Colonoscopy showed a 5-cm-sized ulceroinfiltrative lesion in the sigmoid colon and a polypoid lesion in the rectum. Subsequent histological examination confirmed the diagnosis of adenocarcinoma and high grade tubular adenoma with focal carcinoma in situ transformation, respectively. Incisional biopsy of the spinal mass was performed and the histological examination revealed hemangioblastoma.
The patient underwent laparoscopic right partial nephrectomy, laparoscopic anterior resection of the sigmoid colon, and transanal endoscopic operation of the rectum. Histologic examination of the kidney revealed renal clear cell carcinoma (size, 1.9×1.7 cm), Fuhrman nuclear grade 3 (pT1aN0M0; stage I). In the sigmoid colon, gross pathology showed an ulcerofungating tumor (size, 4.2×3.6 cm) and 3 of 23 examined regional lymph nodes were metastasis. Subsequent histological examination confirmed typical features of colon adenocarcinoma (pT3N1bM0; stage IIIB). The rectum specimen was confirmed to be a 0.5-cm-sized well differentiated adenocarcinoma (pT1N0M0; stage I). The postoperative CEA level was 4.24 ng/mL.
Regarding clinical decision processes, we initially suspected VHL disease. Subsequently, polymerase chain reaction (PCR) sequencing analysis of VHL gene showed p.Glu70Lys (c.208G > A) mutation in exon 1, confirming the diagnosis of VHL disease (Fig. 3). Result of endocrine hormonal evaluation for pheochromocytoma was within normal range.
His two sons were screened for VHL disease using PCR sequencing analysis, and one progenity showed p.Glu70Lys (c.208G > A) mutation in exon 1, identical to his father. He underwent ophthalmic examination, abdomen-pelvic computed tomography scan and blood test for catecholamine metabolites, but there were no abnormal findings.
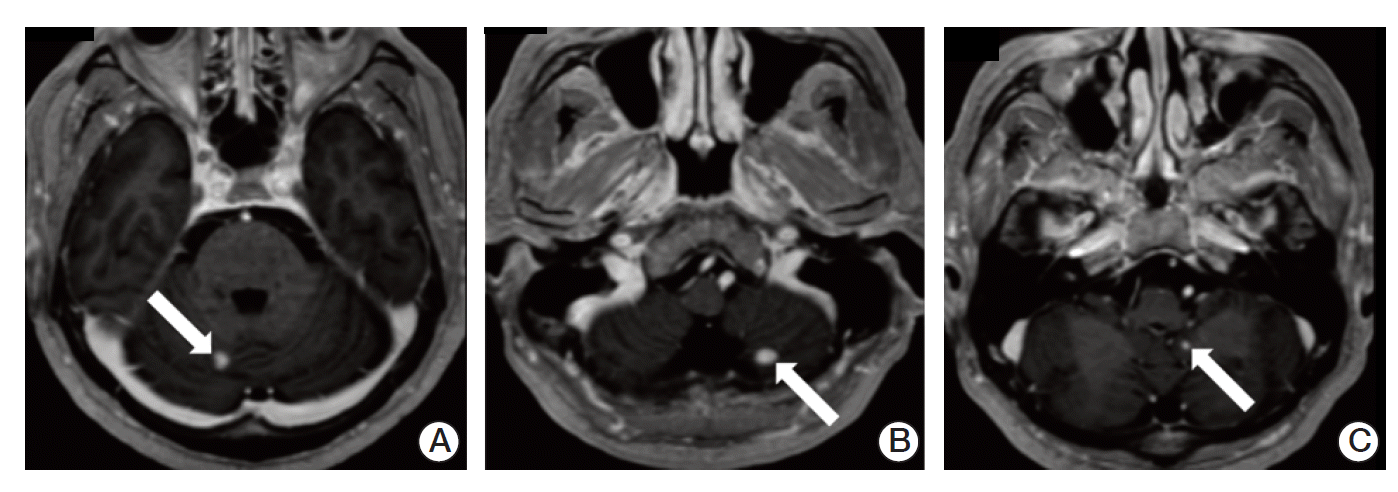
After 3 weeks from discharge, he was admitted to hospital complaining of worsened gait disturbance and numbness of both soles. Brain MRI showed three enhancing lesions in bilateral cerebellar hemispheres and left cerebellar tonsil, suggesting hemangioblastomas (Fig. 4). The patient underwent gamma knife surgery in cerebellar lesions and palliative radiotherapy to T9-S1 spine with a total of 45 Gy in 25 daily fractions.
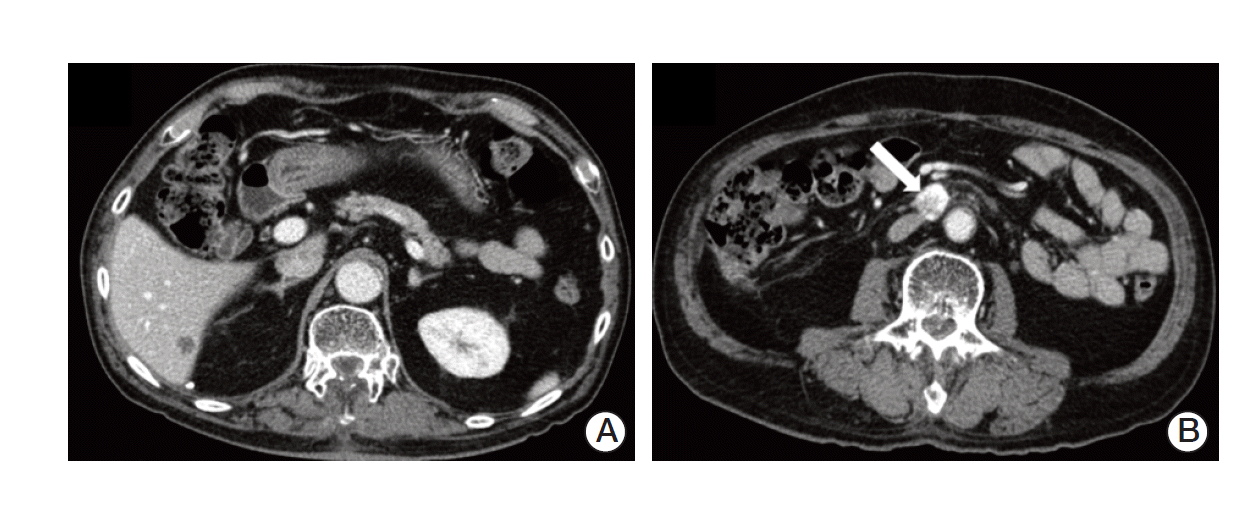
After one month, follow-up imaging studies showed stable lesions in the brain and spine, but there was a newly developed hepatic metastasis in S6 and an enlarged aortocaval lymph node (Fig. 5). Concurrent serum CEA level increased to 19.54 ng/mL. The multidisciplinary team decided to perform surgery, laparoscopic wedge resection of liver and excision of aortocaval lymph node. Histologic examination of liver revealed a 1.6×1.2-cm-sized metastatic adenocarcinoma, moderately differentiated, clinically from colon. Pathologic finding indicated that the aortocaval lymph node was metastatic carcinoma, favoring renal clear cell carcinoma. He is on regular follow-up in the outpatient clinic and imaging studies show no evidence of recurrence and CEA level is stabilized within normal range.
Discussion
VHL disease is a familial multisystem syndrome associated with germline mutation of the VHL tumor suppressor gene at chromosome 3p25. It occurs in approximately 1 in 36,000 live births [2]. The diagnosis of VHL can be made clinically if a single VHL-associated tumor (that is a hemangioblastoma, pheochromocytoma, or renal clear cell carcinoma) occurs in an individual with a family history of VHL. The diagnosis can also be made clinically in the absence of a family history if a second VHL associated tumor occurs in an individual with hemangioblastoma (that is two or more hemangioblastomas or both a hemangioblastoma and a pheochromocytoma or clear cell renal carcinoma). As approximately 20% of VHL disease results from de novo mutations, the absence of a family history in no way excludes the diagnosis [4]. The most common lesions are CNS (retinal and cerebellar hemangioblastoma), kidney (renal clear cell cell carcinoma and renal cyst), adrenal gland/paraganglia (pheochromocytoma/paraganglioma), pancreas (neuroendocrine tumors, pancreatic cyst, or serous cystadenoma), and middle ear (endolymphatic sac tumor) [5].
In this case report, we observed the coexistence of three primary tumors at diagnosis: renal clear cell carcinoma and CNS hemangioblastomas, which are known to present in VHL disease, and colorectal adenocarcinomas. Because his eyes were damaged too severely to evaluate for retina condition, we presumed that he might have had retinal hemangioblastoma in the past. To date, only one case of colorectal carcinoma in a patient with VHL syndrome has been described [6].
The mechanism of VHL syndrome in tumorigenesis is hypoxia inducible factors (HIF1 and HIF2). Either hypoxia or absent or inactive VHL protein results in stabilization of HIF1 and HIF2, which then acts as a transcription factor initiating cellular cascade for hypoxic response. This hypoxic response affects glucose uptake and metabolism, angiogenesis, extracellular matrix formation, chemotaxis and cell proliferation, and then activates downstream targets, including vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), and transforming growth factor α [7].
Several studies offer evidence suggesting that down regulation of VHL provides a proangiogenic impulse for development of colorectal cancer. Xue et al. [8] suggested that chronic increases in expression of HIF in colon initiate protumorigenic signaling. In addition, in the progression from colorectal adenoma to carcinoma, VEGF plays an important role in formation of new capillaries to increase oxygen and nutrient demands [9].
Nevertheless, in this case report, we have weight to explain that synchronous colon and rectal adenocarcinomas developed sporadically in a patient with VHL disease. In Korea, colon cancer is the third most common cancer overall, and the second most common cancer in men [10]. In the general population, the coexistence of primary malignancies of colon and other organs is difficult to assess because the etiology is complex, including both genetic and environmental risk factors.
Early detection and proper intervention currently offer the best hope for minimizing the burden of VHL disease in order to prevent avoidable morbidity and mortality. Either surgical resection or radiation therapy has a role in management of hemangioblastoma in CNS. However, no randomized clinical trials comparing these approaches, or are any randomized trials addressing the optimal timing of therapeutic intervention have been reported. The principle advantage of radiation therapy is that it is noninvasive and does not carry the risks of conventional surgery, such as difficulty to approach, infection, anesthetic complication, stroke, or neurologic deficits in CNS. In particular, in patients with VHL disease, multiple hemangioblastomas tend to develop over time within both the brain and spine, often necessitating multiple operations over a patient's lifetime. Even though these operations take successfully, the patients show gradual disability. In such cases, radiation therapy may be a much safer option [11]. In our case, the patient underwent multiple steps of surgery and radiotherapy. In particular, hemangioblastomas in CNS are controlled safely under radiation therapy regardless of symptoms.
There is no effective therapeutic agent in VHL disease. A small number of clinical trials were conducted, and further study will be needed. For example, Kim et al. [12] reported on sunitinib, tyrosine kinase inhibitor, which inhibits the VEGF and PDGF receptors, and is administered in patients who had metastatic renal cell carcinoma with complicating VHL disease, and it prolonged the duration of response time. As early detection and intervention in time is important for VHL patients, routine surveillance and a multidisciplinary appr-oach due to multisystem involvement are needed. Thus, all VHL patients and relatives who are at risk should be entered into comprehensive screening programs at special referral centers (for example see Table 1 [13]).