Introduction
For many years, chemotherapy, radiotherapy, and surgery have been the mainstay treatment for various forms of human cancer. Despite providing variably effective treatments in many cases, the risk of cancer recurrence due to acquired chemoresistance through different mechanisms, and the significant damage to the surrounding healthy tissue have remained as major hurdles in cancer therapy [1].
In recent years, many components of the immune system have been shown to be effective in restriction of tumor growth and/or tumor elimination, placing immunotherapy on center stage as a biological approach to cancer therapy. In the effort to combat cancer, researchers have utilized diverse strategies, including adoptive transfer of activated T cells and antigen presenting cells (APCs) [2,3], and development of monoclonal antibodies (Abs) against tumor antigens (Ags) [4]. However, these strategies have not been completely successful in control of tumor growth or its eradication. Tumor cells have the ability to evade the adaptive and innate immune defense responses through changes in their surface Ags and synthesis of APC suppressive factors leading to inhibition of T-cell responses [5].
Dendritic cells (DCs) are among the most potent APCs and have a superior capacity to recognize, engulf, and process Ags, be it of foreign origin or mutated host cells, and finally present them to T cells. Depending on the nature of the Ag, the maturation state of DCs and their ability to express costimulatory and/or inhibitory molecules within their microenvironment, DCs play a significant role in modulating the immune response by selective activation and differentiation towards Th1, Th2, Th17, and/or Treg subsets. This overall immunomodulatory capacity makes DCs an attractive candidate for cancer immunotherapy [2].
Significant advances have been made in the development of DC-based cancer vaccines. In many of these vaccines ex vivo–generated DCs are loaded with specific tumor Ags (in the absence of suppressive tumor–associated Ags) and then put back into the host [2]. Several strategies have been developed to maximize the efficiency and longevity of Ag presentation by ex vivo DCs, including simultaneous loading of microbial components with tumor Ags, triggering costimulatory molecules and their ligands on the surface of DCs, and Th1 cytokine gene transfer to DCs [6]. Application of any given single approach per se does not warrant the optimum tumor cell killing capacity and combinations of two or more DC activating strategies are often required [7].
An alternative approach to augment the immunogenicity of tumor Ags and achieve more powerful DC-based cancer vaccines is the use of carrier proteins linked to the tumor Ag [8]. These carrier proteins, also called third-party Ags, boost the CD4 and CD8 responses against the tumor Ag. Timmerman and Levy [9] were the first to demonstrate that loading DCs with tumor Ags linked to an immunogenic protein (keyhole limpet hemocyanin, KLH) significantly increased the antitumor immune response [9]. In another study, they showed that in tumor-bearing mice immunized with idiotype protein-loaded DCs, the anti-idiotype immune response only occurred when the KLH-linked Id protein was used [10]. Millard et al. [11] also reported that DCs pulsed with KLH-linked peptide produced a much stronger peptide-specific immunogenic response compared to the peptide alone group. They also demonstrated that KLH significantly increased the antitumor capacity of DC vaccines and their ability to induce tumor specific cytotoxic T cells through activation of T helper lymphocytes [11].
Our laboratory demonstrated that coupling of a helper Ag with the target tumor Ag increased the efficiency and stability of Ag presentation by DCs and elicited more robust immunogenic responses against both the target tumor Ag and the helper Ag [12]. This phenomenon, which we called a “mutual helper effect,” generates better Ag-specific immune responses against both Ags. Other studies demonstrated that the enhanced tumor-specific Ag presentation was not limited to the inclusion of KLH alone, and many other foreign Ags, even less immunogenic ones were capable of producing powerful DC-based vaccines in normal mice [13,14].
In this work, we attempted to determine whether treatment of a murine model of colon cancer with DCs copulsed with ovalbumin (OVA) (as the helper Ag) and a gp70-derived peptide (AH1) could inhibit tumor growth and induce a potent Ag-specific immune response.
Materials and Methods
1. Animals
Eight- to 12-week-old BALB/c mice were obtained from the Pasteur Institute, Iran. Animals were housed and maintained under optimal light, temperature, and humidity conditions with free access to food and water. All procedures were approved by the Animal Care Ethical Committee at Avicenna Research Center.
2. Cell line
CT26 mouse colon carcinoma cells were purchased from the Pasteur Institute, Iran, and used for this study. These cells express gp70, a murine endogenous tumor Ag. The AH1 peptide of this Ag has also been identified as a cytotoxic T lymphocyte (CTL)–epitope capable of inducing tumor specific CTL immune responses [13]. CT26 cells were routinely maintained in RPMI-1640 medium (Gibco, Karlsruhe, Germany) containing 10% (v/v) fetal bovine serum (FBS; Gibco) and 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO) in 25 cm2 and 75 cm2 flasks (SPL, Pocheon, Korea).
3. Generation of AH1 peptide-specific antibody
Generation of anti-AH1 polyclonal Ab was performed according to a method described elsewhere [15]. In brief, KLH-conjugated AH1 peptide (amino acid sequence, SPSYVYHQF) was injected in conjunction with IMMACELL (Stockholm, Sweden) and Freund adjuvant (Sigma-Aldrich) into an 8-week-old New Zealand rabbit once per week for 3 consecutive weeks and a booster injection was administered 1 month later. Blood samples were collected before the immunization and 1 week after each injection. Sera were separated and stored at –20°C. Specific anti-AH1 polyclonal Ab was purified using affinity chromatography and its purity was checked by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The immunoreactivity of the hyperimmune serum and purified Ab against AH1 peptide was confirmed by enzyme-linked immunosorbent assay (ELISA).
4. Immunofluorescence analysis of gp70 expression in CT26 cells
Expression of gp70 protein in CT26 cells was determined using the above-mentioned purified anti-AH1 Ab according to the protocol, which is described elsewhere [16]. Briefly, cultured CT26 cells were detached by trypsinization, washed, cytospun onto microscope slides, and fixed with ice cold acetone. After blocking endogenous biotin (Dako, Glostrup, Denmark) and the hydrophobic sites (5% normal sheep serum), purified anti-AH1 Ab (4 μg/mL) was applied overnight at 4°C. Cells were then sequentially incubated with biotinylated sheep anti-rabbit IgG (Avicenna Research Institute, Tehran, Iran) and FITC-conjugated streptavidin (Biosource, New York, NY). The nuclei were stained with 7-AAD (Sigma-Aldrich) for 5 minutes. Cells stained with primary Ab, which had been adsorbed with immunizing peptide or pre-immune rabbit IgG, served as the reagent negative control. Fluorescent signals were visualized under an immunofluorescence microscope (BX50, Olympus Optical Co., Tokyo, Japan).
5. Western blot analysis of gp70 expression in CT26 cells and tumors
Expression of gp70 protein in CT26 cells and tumors was determined using the purified anti-AH1 Ab mentioned above. In brief, CT26 cells and tumor tissues were homogenized on ice for 1 hour in a lysis buffer containing NaCl (100 mM), Na4P2O (20 mM), glycerol (1% v/v), Tris (10 mM; pH 7.4) (all from Gibco), Triton X-100 (2% v/v), EDTA (1 mM), NaF (1 mM), and SDS (0/1 % w/v) (all from USB, Cleveland, OH). Subsequently, the lysates were centrifuged, and the supernatants were collected, and stored at –20°C until analysis. Total protein concentration was determined using a BCA kit (Pierce, Rockford, IL) according to the manufacturer's instructions. Lysates were subjected to SDS-PAGE electrophoresis followed by protein transfer onto the PVDF membrane (Millipore, Bedford, MA). Membranes were then blocked (5% skim milk) and incubated with anti-AH1 Ab at 4°C overnight. After washing, the membranes were incubated in horseradish peroxidase–conjugated sheep antirabbit IgG (Avicenna Research Institute) for 45 minutes. AH1 was detected using an ECL Western blot substrate kit (GE Healthcare, Uppsala, Sweden) [17].
6. Isolation and pulsing of DCs
DCs were isolated from spleen of BALB/c mice according to the previously described protocol [18]. Briefly, the spleens were digested with an enzyme blend containing collagenase and DNase, and low density mononuclear cells were separated using Nycodenz density gradient media. Thereafter, DCs were purified by magnetic assisted cell sorting using mouse Pan-DC (Miltenyibiotec, Bergisch-Gladbach, Germany) according to the manufacturer’s instruction. The purity of isolated DCs was tested using flow cytometry analysis of CD11c expression and was shown to be more than 90%.
Splenic DCs were pulsed with a mixture of OVA (Sigma-Aldrich) (100 μg/mL) and AH1 peptide (10 μg/mL) Ags in RPMI+10% FBS at 37°C overnight. Non-adherent mature DCs (DC-Pep-OVA) were then collected and used for injection of mice. DCs pulsed with AH1 (DC-Pep), those incubated in the absence of AH1 (DC-alone), and DCs loaded with OVA and P15 (10 μg/mL) were used as controls.
7. Tumor cell inoculation
CT26 cells were used for tumor induction in mice. Trypsinized CT26 cells were harvested and washed, and their density was adjusted to 5×105 cells/100 μL phosphate buffered saline (PBS). Mice were given subcutaneous injections into the right flank (100 μL). The day of the inoculation was considered day 0 in all groups.
8. Immunization protocols
Tumor-bearing mice were immunized separately with AH1-pulsed DCs (DC-Pep group) and AH1+OVA-pulsed DCs (DC-Pep-OVA group) by injection of 6×105 cells/100 μL PBS into the tumor site. Mice injected with spleen-derived naive DCs (DC-alone group) or with PBS alone (tumor group) served as control. To determine whether the observed responses were specific to the tumor-derived peptide (AH1), we included another control group: DCs pulsed with OVA plus P15, as an irrelevant peptide derived from Micobacterium tuberculosis [12]. Overall, animals were divided into five groups according to the type of injection they received (Table 1). The immunizations were administered on days 3 and 10 following tumor inoculations. In the DC-Pep-OVA group, we observed no sign of toxicity, including fur loss, abnormal behavior, signs of allergy or gross abnormalities in liver, kidney, lung, and gastrointestinal tract.
9. Tumor volume measurement
To determine the effect of DC vaccination on tumor development, local tumor growth was evaluated by measuring two perpendicular diameters of each tumor using a caliper. Thereafter, the volume of the subcutaneous tumor was estimated using the following formula: (short diameter)2×long diameter×0.52. Tumor growth was measured at 3-day intervals for 42 days. On day 42, mice were sacrificed by cervical dislocation and processed for further analysis.
10. Proliferation assay
Splenocyte proliferation assay was performed using a previously described method [19]. Briefly, on day 42 following tumor cell inoculation, splenocytes from mice were harvested by injection of RPMI-1640 medium into the excised spleen and crushing through a mesh. Mononuclear cells were separated by Ficoll density gradient medium. A total of 4×105 splenocytes/mouse from all vaccination groups were plated in 96-well plates (Greiner, Nürtingen, Germany) containing Click’s culture medium (Gibco). Cells were incubated at 37°C and 5% CO2 in the presence of OVA (20 μg/mL), AH1 peptide (20 μg/mL), or PBS for 72 hours, followed by addition of 3H-thymidine (1 μCi/mL; GE Healthcare, Stockholm, Sweden) and incubation for another 18 hours. The cells were harvested, transferred onto glass fiber filters (Whatman, Maidstone, UK), and the level of 3H-thymidyne incorporation was determined using a scintillation counter (Wallac-1410, LKB, Ann Arbor, MI). The assay was performed in triplicate for each sample.
11. Survival of immunized mice
To determine whether DC immunization of tumor-bearing mice prolonged their survival, a separate group of animals underwent the same tumor inoculation and subsequent DC immunization protocol, as shown in Table 1. The survival of these mice was monitored at 7-day intervals and the survival rate was expressed as the percentage of live mice per treatment group.
12. Statistical analysis
Quantitative data from five or seven separate experiments per each control and test group were analyzed using Graph-Pad Prism ver. 6.00 for Windows, GraphPad Software (http://www.graphpad.com; La Jolla, CA), and reported as mean±standard deviation. The statistical significance of differences was determined using Mann-Whitney and/or Kruskal-Wallis non-parametric tests. Log-rank (Mantel-Cox) test was used for comparison of the survival curves of various vaccination groups. p-values less than 0.05 were considered statistically significant.
Results
1. Verification of gp70 expression in CT26 cells and tumors
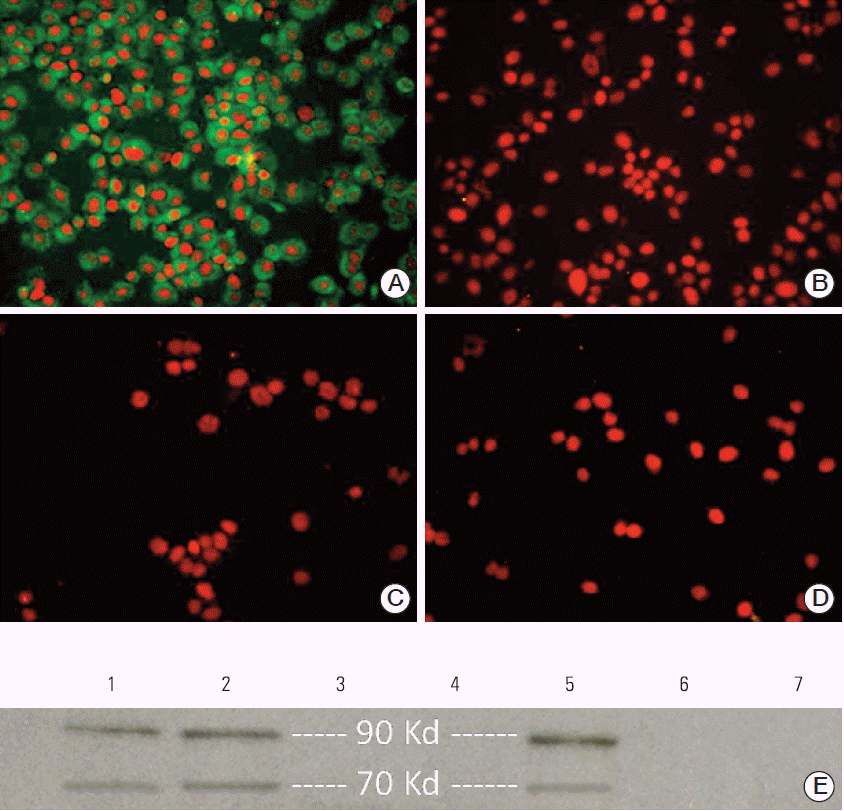
We first generated specific anti-AH1 Ab for assessment of gp70 expression in CT26 cells. Both hyperimmune serum and purified Ab exhibited excellent reactivity with the immunizing peptide as determined by ELISA (data not shown). Expression of gp70 protein in CT26 cells was confirmed by immunofluorescence. As shown in Fig. 1A, anti-AH1 Ab reacted positively with CT26 cells, confirming the presence of gp70. No reactivity was detected in negative controls (Fig. 1B-D). Next, the expression of gp70 protein in CT26 cells and tumors was confirmed by Western blot analysis using our polyclonal anti-AH1 Ab. Muscle tissues from BALB/c mice and 4T1 cells were used as cell negative controls, and rabbit IgG served as the reagent control. Western blot analysis clearly identified the presence of two 70-kDa and 90-kDa protein bands indicative of the presence of gp70 and gp90 (the precursor of gp70 [20]) Ags in both CT26 cells and tumor lysates. No specific bands were identified in negative controls (Fig. 1E).
2. Effect of DC immunotherapy on tumor volume
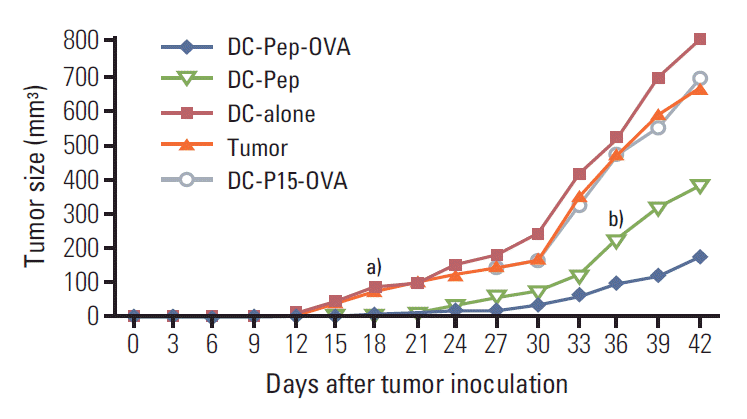
To determine whether DC treatment had any impact on tumor growth, the volume of tumors was measured before and at 3-day intervals after DC vaccination in different treatment groups. In non-vaccinated mice, tumors were visible on day 9, while in the case of DC-alone and DC-P15-OVA groups, tumors were measurable with a 3-day delay on day 12. Of particular interest, vaccination with either DC-Pep or DC-Pep-OVA resulted in delayed onset of visible tumor formation, so that no tumor was observed until days 18 and 21 for DC-Pep and DC-Pep-OVA groups, respectively. From day 18, significantly smaller tumor volume was observed in mice immunized with DC-Pep or DC-Pep-OVA as compared to other control groups (p ≤ 0.05) (Fig. 2). In addition, significantly smaller tumor size was observed in mice receiving DC-Pep-OVA compared to those injected with DC-Pep on day 36 and beyond (p ≤ 0.05). The rate of tumor growth followed an increasing trend in all groups. Nonetheless, in contrast to all other groups, in the DC-Pep-OVA group the tumor size did not grow with a steep slope (Fig. 2).
3. Splenocyte proliferation response in DC immunized mice
As a measure of post-immunization proliferation response to various regimens of Ag loading onto DCs, 3H-thymidine uptake by splenocytes from different treatment groups upon in vitro Ag challenge was quantified. Freshly isolated splenocytes from each mouse were stimulated with either AH1 peptide or OVA and proliferation values were determined. Splenocytes from mice vaccinated with DC-Pep-OVA showed a significantly more robust proliferation response compared to other groups (Fig. 3). In particular, in vitro challenging of the DC-Pep-OVA group with tumor peptide (AH1) showed a significantly higher proliferation response than that of the DC-Pep group. In addition, upon stimulation with AH1, the proliferation rate for the DC-P15-OVA group was extremely low, so that it was comparable to that of the negative control groups (i.e., tumor and DC-alone).
4. Effect of DC immunotherapy on survival rate
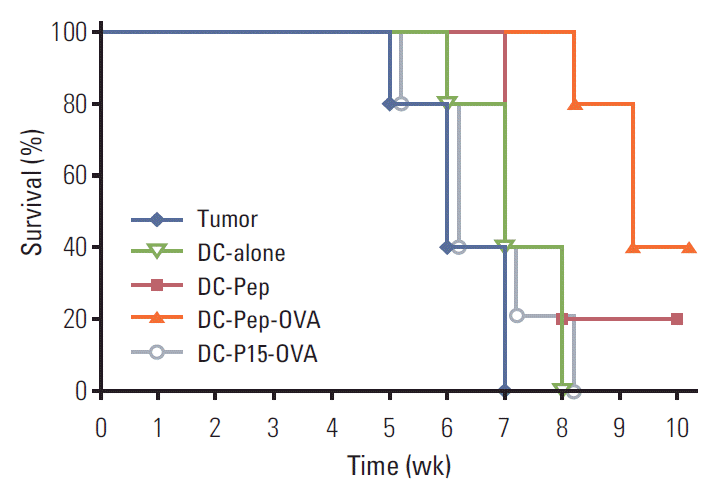
To examine the ultimate efficacy of various DC-based antitumor treatments, the survival rate of each group was further evaluated. Post-immunization survival rate in tumor-bearing mice was presented as the percentage of live mice per treatment group. The population of mice that were not immunized at all, mice immunized with naive DCs, and those injected with DC-P15-OVA showed low survival rates, so that these mice developed lethal tumors. Importantly, immunization of tumor-bearing mice with DC-Pep-OVA significantly prolonged their survival in comparison with mice vaccinated with DC-Pep (p ≤ 0.05) (Fig. 4).
Discussion
Development of efficient methods to cure neoplasm or inhibit its progression has long been at the forefront of tumor immunologists’ attention. In this regard, targeted immunotherapy using DCs has attracted significant interest. In particular, numerous studies have attempted to fully exploit the potential of potent ex vivo–generated antitumor DCs in order to elicit efficient T-cell responses [21]. In our previous study, we showed that the presence of a helper Ag next to the target Ag during pulsing of DCs could enhance the proliferation rate of CD8 T cells specific to both the helper and target Ags, a phenomenon we termed “mutual helper effect” [12]. In this study, elucidate the in vivo effectiveness of this approach against tumor, we vaccinated a murine model of colon cancer with DCs pulsed with the combination of a helper Ag (OVA) and a tumor Ag (AH1). Readout systems including tumor size, survival rate, and tumor Ag-specific T-cell proliferation were employed in order to evaluate the efficacy of DC vaccination.
The main vaccination group consisted of tumor-bearing mice vaccinated with DCs loaded with both AH1 and OVA (DC-Pep-OVA), while the target control group consisted of tumor-bearing mice inoculated with DC-Pep. Tumor size in mice treated with DC-Pep-OVA was significantly smaller compared to that in the DC-Pep group. More importantly, a significantly higher survival rate was observed in the former group. In addition, high tumor volumes and mortality rates in mice treated with DC loaded with OVA plus an irrelevant peptide (e.g., P15) confirmed the formation of responses specific to the tumor Ag, AH1. Together, these results demonstrated that OVA could function in vivo as a helper Ag with the ability to augment the efficacy of tumor Ag-loaded DCs.
In a somewhat similar study, Casares et al. [13] reported that protective injection of normal mice with a combination of an OVA-derived peptide (as the helper Ag) and AH1 (as the tumor Ag) could induce marked tumor protection against challenge in a CT26 cell line. However, there are several major differences between that study and the current work: 1) they did not use DCs for immunization and instead tumor Ag and the helper molecule were injected directly into mice; 2) they used an OVA-derived peptide instead of the intact OVA molecule used in our study; 3) coadministration of OVA and AH1 was used as a protective rather than the therapeutic antitumor modality employed in the current study. Nonetheless, their overall result, similar to our work, supported the idea that an unrelated but proper Th1 molecule could be used as a helper Ag for eliciting tumor-specific CTL responses. The potential advantage of our study over the aforementioned work is that regarding the ability of DCs to present Ags to CD8 T cells, stronger antitumor responses could be provoked upon utilization of Ag-loaded DCs rather than injection of Ags in the absence of DCs [9]. In line with this assumption, in another section of the above-mentioned study, Casares et al. [13] used the combination of AH1 and OVA (again in the absence of DCs) for treatment of tumor-bearing mice, as a therapeutic rather than a protective approach. According to their results, the combination of OVA and AH1 did not prevent tumor growth. In addition, based on the results obtained from the same therapeutic approach, immunization with AH1 alone had no protective effect against challenge with CT tumor cells, while here we showed that, although not significantly, tumor-bearing mice vaccinated with DC-Pep (AH1) showed better survival than those treated with DC-alone or not treated at all.
In another section, we obtained splenic cells from tumor-bearing mice and assessed their proliferative capacity upon in vitro stimulation with either AH1 or OVA. According to the results, splenic cells of mice vaccinated with DC-Pep-OVA showed the highest proliferation rate, which further verified the helper role that OVA could play in enhancement of AH1-specific immune responses. In our previous in vitro study, we showed that injection of DCs loaded with OVA (helper Ag) plus the target Ag could elicit secretion of high levels of interferon γ together with robust proliferative responses of CD8 T cells specific to both Ags [12]. Eriksson et al. [22], using DCs pre-loaded ex vivo with cholera toxin (CT)–conjugated tumor Ag, accomplished induction of efficient CTL-mediated antitumor responses both in vitro and in vivo. In that study, CT functioned as both an adjuvant and a helper protein that was able to elicit non-specific immune responses and facilitate the uptake and presentation of the tumor Ag by DCs [22]. Taken together, these results demonstrate that through utilization of a potent helper Ag, one might be able to promote robust specific immune responses against the Ag of interest (e.g., tumor Ag).
The type of helper Ag and the method used to combine it with the Ag of interest are among the factors that can influence the final results of a DC-based antitumor modality. Accordingly, in an immunotherapeutic study, it was indicated that utilization of a Th0- or Th2-inducing helper Ag, would in fact serve the needs of tumor cells, rather than prevent tumor growth [13]. In the current study, we did not determine the type of T cell–mediated responses; however, as mentioned earlier, in our previous study we observed that OVA-pulsing of DCs would shift T-cell responses towards Th1 [12]. In this regard, Eriksson et al. [23], in two DC-based studies, reported that conjugation of the helper Ag to the target Ag could lead to more pronounced antitumor responses as compared to simply mixing the Ags [24]. Hence, as related to the current study, even more potent antitumor responses might have been achieved upon loading of DCs with a conjugated form of OVA and AH1 rather than a simple mixture of them.
Conclusion
Collectively, our results indicated that co-administration of OVA (a helper Ag) with AH1 (tumor Ag) to DCs could effectively inhibit tumor growth and improve the survival rate as well as Ag-specific T-cell proliferation; this effect seems more pronounced considering the point that simple loading of DC with AH1 alone was less effective. Nonetheless, as mentioned earlier, administration of one therapeutic modality per se would not suffice to overcome the multifaceted escape mechanisms utilized by tumor cells. Therefore, in our future studies we plan to use the helper Ag approach in combination with other related and effective modalities such as adenoviral-mediated overexpression of Th1 cytokines in DCs.