Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare hematologic malignancy that typically involves the skin, lymph nodes, the peripheral blood and the bone marrow. This tumor expresses CD4 and CD56 with no obvious evidence of B- or T-cell differentiation. Since Adachi et al. (1) first described CD4+/CD56+ hematodermic neoplasm in 1994, several individual cases or small groups of cases of this disease have been reported. The prognosis of BPDCN is known to be extremely poor. Relapse frequently occurs within 6 months or less from the initiation of chemotherapy and most patients experience rapid progression that is refractory to conventional chemotherapy. We report here on a case of advanced BPDCN that was initially misdiagnosed as cutaneous lupus erythematosus.
Case Report
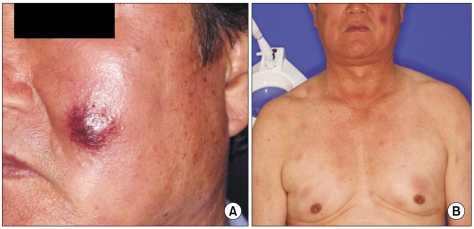
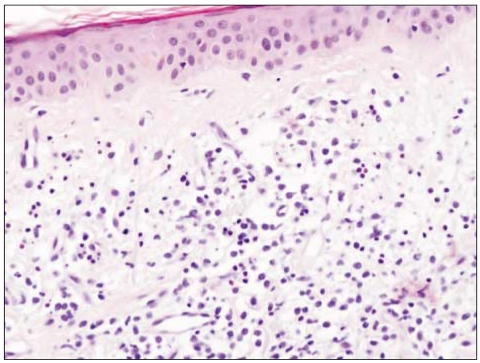
A 67-year-old man with a past history of gout had complaints of non-itching painless skin lesion on the back and it was spreading to the whole trunk, face, arms and thighs for the previous 3 months. This lesion initially presented as a dusky erythematosus patch with indurations on the dorsal area, and it eventually grew and spread. Skin examination showed multiple hyperemic nodules and plaques on the face and trunk (Fig. 1). The antinuclear antibody was negative. A biopsy specimen taken from the facial skin lesion revealed atrophy of the epidermis, a flattened stratum malpighii, small numbers of necrotic keratinocytes and eosinophilic swelling of the dermal collagen fibers, as well as a perivascular and interstitial lymphohistiocytic infiltration in the entire dermis (Fig. 2). On the basis of positive imunophenotyping for CD3 and CD20, the negative imunophenotyping for CD56 and the presence of a mixed reactive lymphocytic infiltration of T and B-cells, a diagnosis of lupus erythematosus was made. A trial of systemic steroid for 3 months improved his rashes.
His rashes developed again on his face and anterior/posterior trunk. Splenomegaly was detected without palpable lymph nodes. The hemoglobin value was 12.2 g/dL, the platelet count was 83,000/µL and the white blood cell count was 22,900/µL with 3% atypical lymphocytes and 7% blasts. The serum levels of lactate dehydrogenase, uric acid and creatinine were increased (1,580 IU/L, 10.5 mg/dL and 1.56 mg/dL, respectively). The marrow biopsy revealed hypercellularity (70%) with diffuse tumor cell infiltration. Marrow aspiration disclosed that blasts comprised 89% of the mononuclear cells and the blasts were of variable size with agranular cytoplasm, and they were positive for CD2, CD3, CD4, CD5, CD7, CD45, CD56, and HLA-DR, and they were negative for myeloperoxidase, terminal deoxynucleotidyl transferase, CD3, CD8, CD10, CD13, CD14, CD19, CD20, CD33, CD34, and CD61. The blood coagulation tests and urine analysis were unremarkable. Cytogenetic analysis revealed a karyotype of 46, XY, add(9)(p24), del(11)(q22).
Repeated biopsy from the left cheek revealed a diffuse lymphoid infiltrate of cells with medium sized nuclei, and the cells were positive for CD4, CD43, CD56 and CD99 (Fig. 3), and this finding was compatible with the diagnosis of BPDCN. Immunostaining for CD3, CD6, CD8, CD20, CD30, T-cell intracellular antigen-1, myeloperoxidase, and terminal deoxynucleotidyl transferase was either weak or negative (Fig. 3). Epstein-Barr virus (EBV) was not detected by RNA in situ hybridization, nor were EBV antibodies detected. A study of T cell receptor gene rearrangement was not performed because the patient did not give informed consent. Computed tomography of the chest and abdomen showed multiple lymphadenopathies in both axillae, the lower neck, the right lower paratracheal area, the lower anterior mediatsinum, the left gastric area, the pericholedocal site and the celiac axis area.
The patient received six courses of multidrug chemotherapy consisting of ifosphamide, methotrexed, etoposide, prednisolone and L-asparaginase, and the patient achieved a partial remission.
Discussion
BPDCN is a rare hematopoietic malignancy, and there has been few data about its incidences. Spanish study (2) reporting the overall incidence of BPDCN among all acute myeloid leukaemia and non-Hodgkin's lymphoma was 0.76% and 0.27%, respectively and Japanese study (3) reporting a 6.33% incidence of this malignancy among natural killer (NK) cell-lineage neoplasm. In Korea, a total of 17 cases of BPDCN have been reported as single cases or as clusters of cases. Among these cases, 4 cases (4) were reported in 2005 and 10 new cases (5) were reported in 2007, and all of these patients died except for three patients who had disease confined to skin.
Since this disease entity was first described (1) in 1994, the nomenclature has changed several times according to the expanded knowledge of this malignancy. The naming of blastic NK cell lymphoma/leukemia was based on the distinct cytology and NK cell origin suggested by the CD56 expression. However, the CD4 positivity and the lack of expressing other NK-lineage markers in most cases cast doubt on its derivation from NK precursors, and hence the French Study Group on Cutaneous Lymphoma (6) called it agranular CD4+/CD56+ hematodermic neoplasm. The World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) consensus classification agreed (7) because of the disease's uncertain lineage. On further study, Chaperot et al. (8) proposed that the plasmacytoid dendritic cell (PDC) might be the normal counterpart of this disease in 2001. On the studies using functional, immunohistochemical and molecular tools, the expressions of the interleukin 3 receptor α-subunit and blood dendritic cell antigen-2 were compatible with a plasmacytoid dendritic cell origin, and the authors of those studies named the disease BPDCN (9). The name BPDCN was enlisted in the 2008 WHO classification of tumors of the hematopoietic and lymphoid tissues. Although the origin of this malady has been defined, the oncogenesis at the molecular level has yet to be clarified.
Histopathologically, BPDCN is characterized by a dense monomorphous infiltration of medium-sized blastoid cells that are positive for CD4, CD56, CD123 and TCL-1. Cota et al. (10) evaluated 33 patients with a proven diagnosis of BPDCN and they found that the cutaneous lesions of BPDCN were quite variable. Most of their cases (80%) presented with nodular/diffuse infiltrates in the entire dermis, fewer cases (20%) presented with relatively small, perivascular or interstitial aggregates of cells, and some cases resembled the picture of a cutaneous inflammatory disorder. The tumor cells in the latter cases were found with reactive lymphocytes, and this made the diagnosis of BPDCN difficult and dicey. In our case, the skin lesion at first biopsy was suggestive of reactive change rather than malignancy. The second biopsy revealed an extensive diffuse lymphocytic infiltration that was positive for CD4, CD43, CD56 and CD99, which is quite a different finding from the first one and it definitely favored the diagnosis of BPDCN. This is certainly a lesson that both clinicians and pathologists should be aware of the atypical presentations of BPDCN from the onset.
Lupus erythematosus is a chronic, inflammatory, autoimmune disease. The histopathologic findings of discoid lupus erythematosus are typified by an interface dermatitis involving the hair follicles and epidermis, and this is accompanied by a moderate to heavy superficial, deep perivascular and periappendageal lymphocytic infiltration. Plasmatoid dendritic cells are known to play an important role (11,12) in the pathogenesis of lupus erythematosus and these cells produce type I interferon. Vermi et al. (13) showed that a cutaneous plasmatoid dendritic cell infiltration is a hallmark of lupus erythematosus. Although it is thought that BPDCN originates from PDCs, there has been no report of BPDCN in the setting of lupus.
BPDCN mainly occurs in older patients, although it can occur at any age, including the pediatric period. It affects males more frequently than females (12) at a proportion of roughly 3:1. It frequently involves multiple sites with a predilection for skin, and this is followed by bone marrow, peripheral blood and lymph nodes. The skin lesions tend to be localized at the onset and then they become multiple or more generalized during the course of disease, and overt leukemia is the usual finding in advanced cases or there is relapse after therapy. Rare cases involve the central nervous system at presentation.
Frequent chromosomal aberrations in various sites have been reported for this disease, including chromosomal losses on 5q, 6q, 12p, 13q and chromosomes 9 and 15, as well as deletion of regions on 4q34, 9p13-p11, pq12-q34 and 13q12-q31. The significance of these chromosomal abnormalities is not yet certain. Our case demonstrated an abnormal karyotype of 46, XY, add(9)(p24), del (11)(q22). The comparative genomic hybridization performed in the 4 cases reported in Korea (5) showed amplification of 11q12.31 in all 4 cases and 12q13.11 in three.
The clinical course of BPDCN is aggressive and generally dire at best, with the median survival time being 12-24 months. Although there is no consensus on the optimal treatment for this neoplasm, a systemic treatment should be started at the outset regardless of whether the disease is localized or disseminated. Several reports described a better prognosis for cases with skin involvement alone. The clinical course in most cases has been characterized by an initial response to chemotherapy followed by relapse and subsequent death. As the outcomes of many therapeutic attempts have been reported to be quite variable, there is no standard treatment for BPDCN. Usually the duration of a response to topical steroid, single agent chemotherapy, radiotherapy and polychemotherapy like CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) or HyperCVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone alternating with high dose methotrexate and high dose cytarabine) has been reported to be brief. Pralatrexate was anecdotally reported to be active as a salvage treatment (14) although its impact on the clinical course looked negligible. In the face of the extremely poor outcome of conventional chemotherapy and radiation therapy, Dalle et al. (15) asserted that only stem cell transplantation can significantly prolong survival regardless of the initial extent of disease. They claimed that stem cell transplantation should be recommended for every eligible patient. Although our patient has shown a partial response to six cycle of chemotherapy, his ultimate prognosis looks grim.
Our case taught us a lesson that BPDCN should be considered in the differential diagnosis of skin rashes that are refractory to treatment. Although we could not identify PDCs in skin on the first biopsy in our case, their accumulation has been described in several inflammatory cutaneous conditions (12) such as systemic lupus erythematosus, psoriasis, lichen planus and contact dermatitis. Given its easy accessibility and relative safety, we advocate that repeated skin biopsy should be performed in every case of unusual skin lesion.