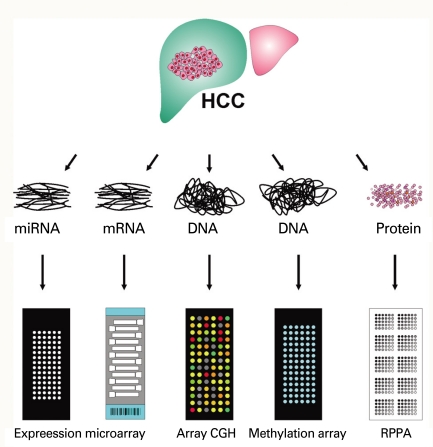
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers in the world, accounting for an estimated 600,000 deaths annually [1]. Although much is known about both the cellular changes that lead to HCC and the etiological agents (i.e., hepatitis B and C infections, alcohol) responsible for the majority of cases, the molecular pathogenesis of HCC is not well understood [2-4]. Moreover, the severity of HCC, the lack of useful diagnostic markers and effective treatment strategies, and the clinical heterogeneity have rendered the disease a formidable challenge in oncology [4,5]. Patients with HCC have a highly variable clinical course [3,6], indicating that HCC comprises several biologically distinct subgroups providing an opportunity for improved classification, identification of novel targets and improved outcomes.
Several clinical classification systems, including the Cancer of the Liver Italian Program, the Barcelona Clinic Liver Cancer system, the Chinese University Prognostic Index, and the Japanese Integrated Staging schema, have been developed and are currently in use [7-10]. However, clinical and pathological diagnosis and classification of HCC remain unreliable in predicting patient survival and response to therapy. The prognostic variability likely reflects a molecular heterogeneity that has not been appreciated from methods traditionally used to characterize HCC combined with a lack of a deep mechanistic understanding of the molecular mechanisms driving disease initiation and progression. Improving the classification of HCC patients into groups with homogeneous prognosis, as well as a more comprehensive understanding of the underlying biology of HCC development at the molecular level, would improve the application of currently available treatment modalities and offer the possibility of new treatment strategies.
Because of the complex nature of cancers such as HCC that are highly heterogeneous at molecular, cellular, tissue, organism, and population levels, conventional "reductionist approaches," which investigate a single gene or protein at a time, are likely to provide only limited insight into the pathological and biological characteristics. Moreover, the rapid advance of technologies that collect large amounts of data from cancer patients or tissues presents another challenge in interpretation and development of core insights into these complex systems. Systems biology, generally regarded as the "comprehensive approach," has been developed to address these issues by blending high-throughput data collection, computational and mathematical modeling, and generation of new hypotheses from emergent properties [11,12]. Emergent properties are those that are not intuitively obvious in the absence of robust and usually mathematical models. In systems biology, large networks describing the regulation of entire genomes, metabolic pathways, or signal transduction pathways are analyzed in their totality at different levels of biological organization. Thus, this approach has been used for generating new hypotheses rather than testing existing hypotheses.
One of the most exciting developments in recent years has been the clinical validation of targeted drugs that inhibit the action of pathogenic gene products such as protein kinases and proteinases [13]. Treatment with these targeted drugs has proven more efficient than conventional therapies in altering the natural history of the disease and reducing mortality for various cancers, including HCC [14-16]. However, molecular characterization of HCC aimed at identifying driver oncogenes (potential therapeutic targets) has lagged in comparison to other cancers. To improve treatment options and reduce mortality for HCC, therefore, it is crucial to develop treatment strategies that can be applied in the near future while improving our understanding of hepatocarcinogenesis.
DNA Copy-Number Alterations in HCC Genome
Since the discovery of aneuploidy, copy number aberrations and genetic rearrangements in cancer [17], cytogenetic approaches have been used extensively to uncover the chromosomal basis for genetic alterations in cancer. The comparative genomic hybridization (CGH) technique was developed in the early 1990s and was the first genomic tool to provide a genome-wide characterization of copy-number changes in cancer [18]. With improvements in microscope and labeling technologies, CGH has become a frequently used tool to examine DNA copy-number changes in cancer and to identify altered expression and function of genes residing within the affected region of the genome. Such genomic loci are believed to harbor either tumor suppressor genes or oncogenes in loci with decreased and increased copy numbers, respectively. Despite limited spatial resolution of CGH mapping, approximately 10 megabasepair (Mbp) for low copy-number gains and losses and close to 2 Mbp for high copy-number amplifications, this technology has uncovered many candidate loci for tumor suppressor genes and oncogenes in HCC. Identification of genomic loci with copy-number aberrations combined with the capacity to identify the genes residing in these loci led to a better understanding of the development of these cancers. For example, increased copy number of the 8q24 region has been reported in many studies, and the most potent oncogene residing in 8q24 is MYC [19-22]. CGH data revealed that gains of chromosomal material were most prevalent in (besides 8q) 1q, 6p, and 17q, and losses were most frequent in 8p, 16q, 4q, and 17p [19,22].
Sensitivity in detecting copy-number variations improved significantly with the emergence of microarray-based technology; in array-based CGH, arrays of genomic sequences such as BAC clones and oligonucleotides replaced metaphase chromosomes as hybridization targets [23]. Coupled with improved annotation of genome sequence data, these technologies are facilitating identification of new genomic loci that are associated with cancer progression.
Microarray-Based Technologies
The genetic or epigenetic basis of complex diseases such as cancer remained largely indefinable until completion of the human genome project and the arrival of new microarray-based technologies that have enabled investigators to describe genetic variation across the entire genome. Completion of the human genome sequence was a crucial prerequisite for cataloging our genetic makeup. However, comprehension of the sequence data alone is not sufficient to decipher the complex physiological processes in play during tumor development.
Microarrays are the technologies most frequently used now to collect data on a global scale from any biological system of interest. Recent advances in our knowledge of the chemistry of oligonucleotides and the availability of genome information helped us to miniaturize northern blots to measure thousands of gene expressions simultaneously. The microarray technology for gene expression is a quantitative assessment of the relative amount of the specific mRNA that is directly related to the biological activity of that particular gene. The amount of the mRNA transcript present in tissues can be measured indirectly after hybridization of a complementary labeled cDNA with a complementary probe that has been previously deposited on its solid surfaces. This array contains a known set of gene sequences (genome). The intensity of each labeled cDNA directly reflects the expression level of its corresponding gene. Microarray assays allow massive parallel data acquisition and analysis. Although parallelism greatly increases the speed of data collection, the massive resulting dataset presents daunting challenges to processing and interpretation.
Microarray-based gene expression profiling studies in a variety of cancers have discovered consistent gene expression patterns associated with pathological or clinical phenotypes, and have identified subtypes of cancer previously undisclosed with conventional technologies [24-26]. This new technology has been used successfully to predict clinical outcomes and survival rates and to identify potential therapeutic targets and prognostic marker genes [27-29].
Application of this technology is not limited to collection of gene expression data from cells or tissues, but extends to identification of DNA copy numbers in cancer genomes, methylation status of gene promoters, single nucleotide polymorphisms associated with cancer risk, protein arrays, and even re-sequencing of whole genomes (Fig. 1). Array-based CGH, in which arrays of genomic sequences are used as hybridization targets, was quickly established as a substitute for conventional CGH [30,31]. The biggest advantage of array-based CGH is the ability to perform copy-number analyses with much higher resolution than was ever possible with conventional CGH, which used metaphase chromosomes as hybridization targets.
Protein microarrays have been developed by adopting the knowledge and technical innovations that have made DNA microarrays possible. The technical aspects of miniaturizing traditional methods, such as western blotting and protein dotting onto nitrocellulose or nylon membranes, were quickly adapted to protein microarray technology. The two approaches for producing protein microarrays are forward-phase protein array (FPPA) and reverse-phase protein array (RPPA). In a forward-phase array, antibodies are immobilized on the surface of slides and each array is incubated with one test sample such as a tissue lysate or serum sample; multiple protein features such as expression and phosphorylation from that sample are measured simultaneously. In contrast, the RPPA format immobilizes an individual tissue lysate in each array spot, and thus an array comprises thousands of different patient samples. Each array is then incubated with one antibody, and a protein feature is measured and directly compared across multiple samples. FPPA (antibody array) is particularly ill suited for tissue-based analysis since it requires a substantial amount of tissue lysate for incubation, thus RPPA (tissue lysate array) is better choice of platform in cancer research [32-35].
Next Generation Sequencing
During the last few years, there have been remarkable advances in DNA sequencing technologies, with the emergence and rapid evolution of massive parallel sequencing or 2nd generation sequencing. These technologies have dramatically reduced both cost-per-base and time of these analyses, making it possible to determine the nucleotide sequence of the human genome [36]. They provide unprecedented opportunities to examine every nucleotide sequence of the DNA from cancer cells and to compare it to that of normal cells to identify the genetic changes that occur during cancer development. Many different platforms have been developed by different companies: Life Technologies (sequencing by Oligonucleotide Ligation and Detection or SOLiD, Carlsbad, CA), Illumina (Genome Analyzer II, San Diego, CA), Roche Applied Science (454 Genome Sequencer FLX System, Indianapolis, IN), and Helicos BioSciences (HeliScope™ Single Molecule Sequencer, Cambridge, MA).
Gene Expression Profiling of HCC
Conventional approaches to the prognostic classification of HCC largely rely on single or multiple clinicopathological variables such as severity of liver impairment or characteristics of the tumor (i.e., size, number of nodules, vascular invasion, distant metastasis, and tumor differentiation grade). However, the utility of existing prognostic factors is limited because they measure tumor differentiation and bulk but do not otherwise characterize and/or measure underlying biological properties that likely dictate clinical outcomes and responses to targeted therapies.
In previous studies [26,37-40], an unbiased analytical approach applied to gene expression data from human HCC identified distinct subtypes of HCC significantly associated with patient survival. These findings suggest that gene expression profiling signatures accurately reflect biological and clinical differences between subtypes of HCC and would be highly valuable in determining patient prognosis. The current clinical challenge is to identify patients who do not derive much benefit from conventional therapies and to offer alternative treatments. If key (or master) regulators (genes, pathways, and/or networks) driving the biology of the tumor can be identified, they might lend themselves to therapeutic exploitation. In this context, it is not enough to rely entirely on gene expression signatures that are indicative of prognosis, since the profiles may fall short of explaining at the molecular level what drives the prognostic difference between subtypes of tumors.
Integration of Multiple Data Sets: Cross-species and Cross-platforms
Although the process of cancer development in humans has differences from that in mice, the similarities are particularly striking [41,42], leading many investigators to exploit the mouse as a model organism for the study of this complex disease. Previous studies provide clues on how to extend gene expression profiling studies beyond the current general practice of collecting massive data from human cancers. In an effort to identify the mouse HCC models that best mimic the human disease, gene expression data from patients were integrated with those from mouse HCC [43]. Gene expression patterns of mouse HCC were obtained from several HCC mouse models. Orthologous human and mouse genes from both datasets were selected before gene expression data were integrated. In analysis of integrated data, gene expression patterns of HCC developed in Myc, E2f1, and Myc/E2f1 transgenic mouse models had the greatest similarity with those of the longer surviving group of humans with HCC, while the expression patterns of HCC in the Myc/Tgfa transgenic mouse model were most similar to those of the poor survival group of humans with HCC. These results suggest that these two classes of mouse models might most closely recapitulate the molecular patterns of the two subclasses of human HCC.
Recent studies demonstrated that Sav1 and Mst1/2 knockout in liver leads to development of HCC [44-47], strongly indicating that MST1/2 and SAV1 are important tumor suppressors in liver. In future studies, therefore, it will be necessary to cross-compare well-defined molecular signatures of these mouse models to those from human HCC to determine the clinical relevance of inactivation of MST1/2 and SAV1 in human HCC. We anticipate that unique molecular identities of each subclass of HCC uncovered by comparative analysis of a genome-wide survey of gene expression from human and animal models will provide new therapeutic strategies to maximize the efficacy of treatments.
Cancer cells do not invent new pathways. They evolved from normal cells by using pre-existing pathways in different ways or by combining components of these pathways in a way that effectively drives tumorigenesis. By mapping and refining pathway maps in developing or normally functioning liver, gene expression profiling studies might provide insight into the connectivity of these pathways in HCC. In a previous study, the gene expression signature unique to rat fetal liver progenitor cells was integrated with those from human HCC in an attempt to determine the fraction of human HCC that shares gene expression patterns with liver progenitor cells [40]. This approach identified a novel subtype of HCC that may arise from hepatic progenitor cells. This new subtype accounts for around 20% of HCC cases examined in this study and is associated with extremely poor prognosis.
Previous studies in diffuse large B-cell lymphoma and T-cell acute lymphoblastic leukemia indicated that the cellular origins of a tumor largely dictate the clinical outcome [24,48], since mitogenic, motogenic, and morphogenic responses as well as the propensity for apoptosis may vary at different stages of normal differentiation. Genes involved in an invasive phenotype (MMP1, PLAUR, TIMP1, CD44, and VIL2) were strongly expressed in the subtype with hepatic progenitor cell features and may account for the extremely poor prognosis. This subtype showed marked activation of AP-1 complex, which is essential for normal hepatogenesis during embryonic development and critical for initiation of HCC development in mice [49,50]. Cancer cells arise from normal cells following accumulation of genetic alterations. One of the most important consequences of this process is the resurrection of pre-existing but dormant signaling pathways that were active during embryonic development [51]. Thus, this finding supports the growing appreciation that signaling pathways that control vertebrate embryonic development are also important in human carcinogenesis.
Searching for Therapeutic Targets
Many studies clearly demonstrated the gene expression signature as a utility that can classify tumors and provide prognostic information [5,26,40,43,52-54]. The current research focus has shifted toward identifying genetic determinants that are components of specific regulatory pathways altered in cancers, potentially leading to the discovery of novel therapeutic targets [4,55-57]. However, selection of relevant candidate genes for further studies from lengthy gene lists generated by gene expression profiling studies is a significant challenge due to the many confounding factors embedded in the gene expression profile data from human cancers. Moreover, since the gene expression profile from patients is only a "snapshot" of gene-to-gene interactions that lacks information on interactive time-dependent changes during tumorigenesis, it is difficult to discriminate the genes (drivers) that drive the tumorigenic process from genes (passengers) whose expression patterns simply reflect loss of organ function and/or degree of differentiation of the cancer cells.
As already discussed, CGH, and more recently array-based CGH analyses, have identified a number of recurrent regions of DNA copy-number changes in many cancers, including frequent DNA copy-number gains at 1q, 8q, and 20q, and losses at 1p, 4q, 8p, 13q, 16q, and 17p in HCC [4]. Some of these genomic loci contain well-characterized and/or putative oncogenes and tumor suppressor genes. Moreover, a number of genes in these regions have been linked to disease pathogenesis and clinical behavior. For example, associations of DNA copy-number aberrations with prognosis have been found for a variety of tumor types, including prostate cancer, breast cancer, gastric cancer, multiple myeloma, lymphomas, and HCC [57-62]. However, some amplified or deleted regions are large, and many of the genes residing in recurrent regions appeared to be silent in their expression in normal tissues as well as in tumors. Moreover, functional validation of genes residing in these loci is impractical when confronted with hundreds of candidate genomic loci. Therefore, there is an inevitable need for development of a new strategy that can overcome the limitations of gene expression data and array-based CGH data.
In a recent study in HCC, investigators tested the possibility that integrating gene expression and gene copy-number data from the same patient cohort would help identify potential driver genes [63]. The results clearly demonstrate that gene copy-number data provided extra prognostic relevance compared to when only gene expression data were available. Integrative analysis of gene expression and gene copy-number data also uncovered 50 potential drivers that are activated by recurrent gene amplifications in HCC and show an association with aggressive tumor types.
Alterations of expression patterns and genomic copy numbers of thousands of genes are fundamental properties of cancer cells. Since the application of high-throughput genomic technologies based on microarray, mass spectrometry, and 2nd generation sequencers for the analyses of cancer inevitably generates many false-positive results, it is almost impossible to select reasonable numbers of candidate genes to be further evaluated as therapeutic targets and/or biomarkers for diagnosis and prognosis. Therefore, it is important to cross-compare and integrate two or more genomic scale datasets (i.e., coding and noncoding gene expression and array-based CGH data or promoter methylation) independently collected from the same patient cohort.
Integration of Multiple-omics
While gene expression and copy-number profiling can provide important information on somatic genetic events during tumor progression, they are unable to provide an effective recapitulation of fluctuating protein-based signaling events that are the direct executors of cellular function. RPPA is a newly developed high-throughput functional proteomic technology [32-35] that provides quantitative analysis of the differential expression of signaling proteins. Moreover, the phosphorylation status of proteins can be detected and measured using specific anti-phospho-protein antibodies. Through the use of these phospho-specific antibodies, it is possible to evaluate the state of entire portions of a signaling pathway by looking at dozens of kinase substrates at the same time through multiplexed phospho-specific antibody analysis.
With RPPA, all samples are spotted at the same time and analyzed with a single antibody, making this method ideally suited for analysis of large numbers of specimens. However, its assessment of signaling pathways is limited by the number of available antibodies, which is far smaller than the number of gene probes in expression microarrays. This limitation of these datasets can be overcome by integrating datasets together during analysis (Fig. 2). Integration of genomic and proteomic data will undoubtedly enhance our understanding of tumor progression by increasing the dimensionality of molecular features. Moreover, identification of driver or contributor genes can be greatly accelerated by integration of more than one dimension of genomic information systems.
Epilogue
The anticipated benefits of the systems biology approach in HCC, which has extremely heterogeneous properties (definite but various etiologies, diverse residual liver function, and heterogeneous tumor biology) would be more reliable determination of prognosis than conventional staging systems, optimized prediction of response to respective treatments, and novel identification of better therapeutic targets. In contrast to other solid tumors, many HCC develop in patients with considerably impaired organ function, which inhibits appropriate therapy and ultimately shortens survival. Therefore, it is crucial to develop a prognosis prediction system that is able to differentiate as well as integrate the impacts of liver function and tumor burden. Several distinct treatment options have been applied to HCC patients [64]: liver transplantation, resection, local ablation, transcatheter intraarterial chemoembolization, and novel targeted therapy sorafenib. Moreover, the indications for respective therapies frequently overlap, and which therapy is optimal is still being debated. Therefore, it will be necessary to develop discrimination systems that would identify the best treatment for an individual patient. Systems biology approaches with genomic and proteomics tools hold promise for personalized medicine in the management of HCC.