Introduction
Melanoma has one of the highest somatic mutational burdens among solid malignancies [1]. In the last decade, a number of important genetic alterations have been identified during various stages of melanoma progression leading to a better understanding and molecular classification of the disease [2]. Unlike the older histological classification, molecular approaches define melanoma as a more heterogeneous and rather complex neoplasm [3]. In 2015, The Cancer Genome Atlas (TCGA) program described the landscape of genomic alterations in cutaneous melanoma in which established a framework for genomic classification into four subtypes: BRAF subtype, RAS subtype, NF1 subtype, and triple wildtype (RAF, RAS, and NF1 wild-type) melanomas [4]. This classification may guide clinical decision-making for targeted therapy such as BRAF inhibitor or tyrosine kinase inhibitor [5], and also offers insights to further personalized therapy.
Like other malignancies, melanoma is a highly heterogeneous neoplasm, composed of subpopulations of tumor cells with distinct molecular and biological phenotypes [6]. Moreover, genotypic and phenotypic analyses of metastatic melanoma cells have revealed that the tumors are more heterogeneous than the primary lesions [7]. In this study, we investigated the mutation status of primary and metastatic melanomas in Korean patients, and their association with clinical features and impact on the outcome of melanoma patients.
Materials and Methods
1. Patients
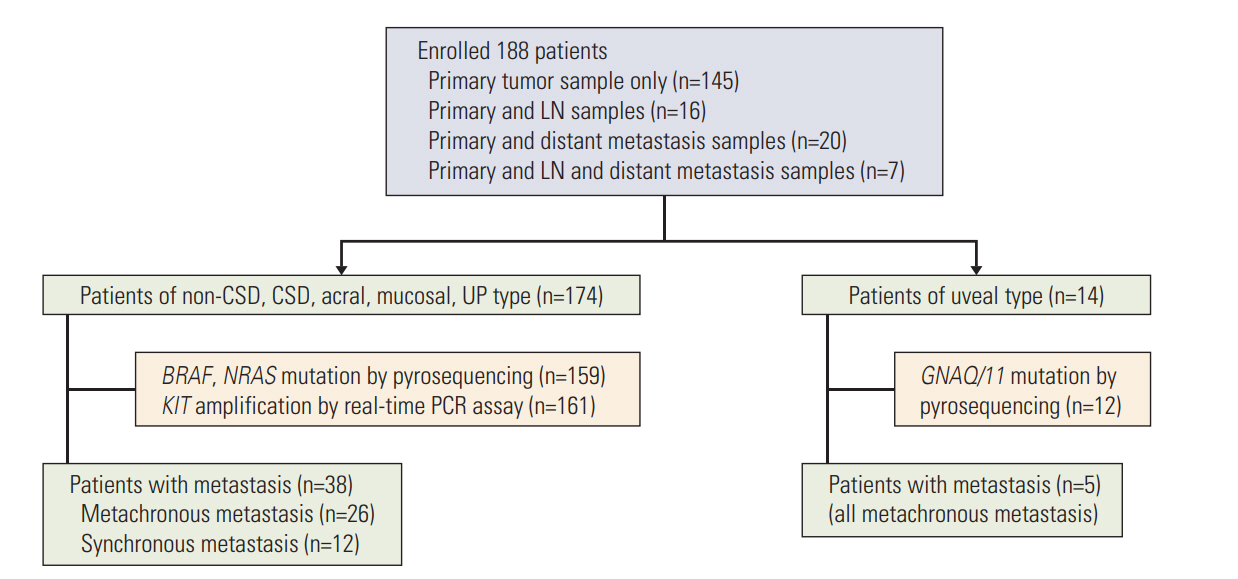
This study included 188 melanoma patients, diagnosed during January 2005 to January 2012, at Yonsei University College of Medicine, Severance Hospital and Yonsei Cancer Hospital in Seoul, Korea (Fig. 1). Among the 188 patients with primary tumors, 43 patients had lymph nodes or distant metastatic lesions also. Clinical data including age, sex, tumor-node-metastases (TNM) stage, tumor thickness (Breslow), ulceration, and survival (follow-up persisted until the missing of follow-up or the death of patients) were collected. The staging was determined according to the American Joint Committee on Cancer (AJCC) guidelines for melanoma at the time of diagnosis.
2. DNA preparation and mutation analyses
Formalin-fixed, paraffin-embedded tissue blocks were retrieved and confirmed as malignant melanoma reviewed by two independent pathologists. Tumor-rich areas (> 80%) were extracted from five paraffin sections of 10-μm thickness containing a representative portion of each tumor block, using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). To detect hotspot mutations, we amplified exons 15 (codon 600) of the BRAF gene, exons 1, 2 (codon 12, 13, and 61) of NRAS gene, and exons 4, 5 (codon 183 and 209) of GNAQ/11 gene by polymerase chain reaction (PCR). The primer sequences are listed in S1 Table. GNAQ/11 gene mutation was only detected in uveal melanoma samples. We performed pyrosequencing using a PyroMark Q24 (Qiagen, Germantown, MD) at room temperature with PyroMark Gold Q24 Reagents (Qiagen) following the manufacturer’s instructions. Sequencing analysis was performed using PyroMark Q24 software ver. 1.0.10 (Qiagen).
3. Real-time PCR assay for KIT copy number
KIT copy number was assessed by quantitative real-time PCR using glyceraldehydes-3-phophate dehydrogenase (GAPDH) as a control gene (The KIT exon 17 primers and GAPDH primers are listed in S1 Table). PCR reactions were done by QuantiTect SYBR Green PCR Kit (Qiagen), with a 20 μL total volume and 100 ng genomic DNA by Rotor-gene 2000 Real-Time Cycler (Corbett Research, Mortlake, Australia). The PCR condition was 1 cycle of 95°C for 15 minutes, followed by 40 cycles of 95°C for 20 seconds, 50°C for 30 seconds, and 72°C for 45 seconds. Relative copy numbers were calculated by the ΔΔCt method, where Ct is the threshold cycle for amplification. For each sample, ΔCt for KIT versus GAPDH was calculated as ΔCt=Ct (KIT)–Ct (GAPDH). The ΔCt value for each experimental test sample was calibrated to a reference pool of human genomic DNA (Promega, Madison, WI), using the formula ΔΔCt=ΔCt (test sample)–ΔCt (reference pool). Relative DNA copy number was calculated using the formula 2-ΔΔCt.
4. Statistical analysis
All the statistical analyses were performed using SPSS ver. 18.0 software (SPSS Inc., Chicago, IL) and MedCalc ver. 12.7.4. Categorical data are described using frequencies and percentages. Continuous data such as age are described using mean±standard deviations or median (range) for normally distributed data. Chi-square test or Fisher exact test was used to differentiate the rates of different groups, and differences in measurement data of two groups were evaluated by unpaired t test or Mann-Whitney test. Survival curves were established using the Kaplan-Meier method and compared by the log-rank test. All statistical analyses were two-sided, and significance was assigned at p < 0.05.
Results
1. Patients and tumor tissue samples
In a total of 188 melanoma patients, the median age was 60 (range, 18 to 89 years) and the sex (male:female) ratio was 1:1.1. The most common subtype was acral type (n=89, 47.3%) followed by non‒chronic sun-induced damage (CSD) type (n=32, 17%) and mucosal type (n=31, 16.5%). CSD type consisted of 18 patients (9.6%) and 14 patients (7.5%) had uveal type, while four patients (2.1%) had the tumors of unknown primary origin (UP) (Table 1). The UP was diagnosed in patients who had initial presentation of melanoma in the lymph nodes, or subcutaneous tissue with no known primary site. Of the total patients, 62.8% (n=118) were localized tumors (stages I and II) and 27.2% (n=70) were advanced tumors (stages III and IV). CSD and uveal type tend to have more localized tumors than the other types (77.8% and 64.2%) while mucosal type was more likely to have the advanced tumors (54.8%) (Table 1).
2. Mutation types and frequencies of melanoma
Recent study of genetic alterations in 333 cutaneous melanomas showed that mutations in BRAF V600 and NRAS Q61 were found in 46% and 24%, respectively. Although other genes, including NF1, HRAS, and NRAS, were highly mutated in cutaneous melanoma, there are no specific hot spots, which are frequently mutated as BRAF V600 and NRAS Q61, in these genes [4]. According to a previous study, KIT amplification was found in 40% of acral melanoma and 31% of mucosal melanoma in Korea [8]. Since our cohort mainly comprised acral (47.3%) and mucosal (16.5%) melanoma, we evaluated genetic status of BRAF (V600) and NRAS (G12, G13, and Q61), as well as KIT amplification for cutaneous melanoma in study. However, uveal melanoma is frequently associated with GNAQ/11mutations and lacks BRAF mutation, NRAS mutation and KIT amplification [9,10]. Therefore, we sequenced GNAQ/11 (R183 and Q209) only for uveal melanoma in this study.
The overall incidence of somatic mutations within the BRAF V600 gene was 17.6%, NRAS 12.6%, and KIT amplification 28.6% in primary melanoma samples. BRAF and NRAS mutations were mutually exclusive as expected. GNAQ/11 mutation was found in 66.6% of the uveal type (Table 2). Compared to other subtypes, non-CSD type and acral type were highly associated with BRAFmutation (41.9%) and KIT amplification (35.8%), respectively. The BRAF V600E mutation was the most common genetic alteration in this study (n=27). BRAF V600K mutation was found in only one patient who was CSD type.
3. Association of BRAF, NRAS mutations, and KIT amplification to the clinical features
In this study, patients with BRAF mutation were significantly younger (median age, 54 years) than those with wildtype BRAF (median age, 60 years) (p < 0.01) (Table 3). Also advanced tumors in stages III and IV (67.8%) were more significantly noticed in patients with BRAF mutation whereas patients with wild-type BRAF were more in stage I and II (68.8%). No such tendency was seen in patients with NRAS mutation or KIT amplification (Table 3).
4. Prognostic significance of BRAF, NRAS mutations and KIT amplification with overall survival of melanoma
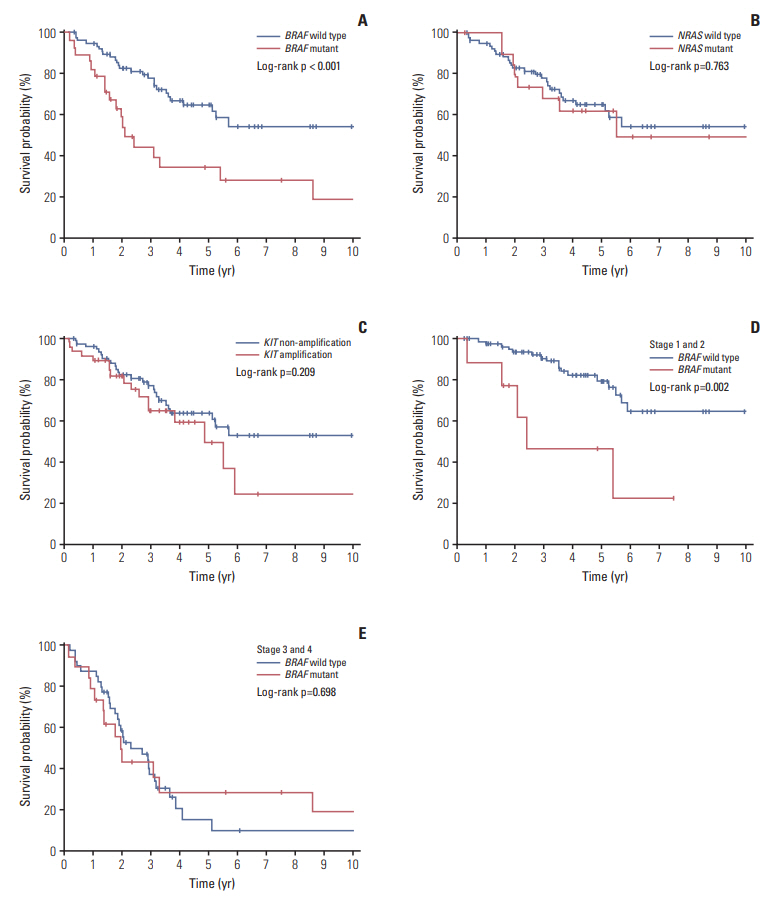
We observed that the median survival time for 28 patients with BRAFmutation (22 months; range, 2 to 158 months) was significantly shorter than that of 80 patients with wild-type patients (39 months; range, 4 to 204 months; p < 0.001) (Fig. 2). To exclude the effect of targeted therapy, including anti-BRAF treatment, on survival of melanoma patients, patient treated with targeted therapy was not included in this study. However, NRASmutation or KIT amplification did not show any significant effect on the survival of melanoma patients. Interestingly, the effect of the BRAF mutation was much stronger in the patients with localized stage tumors (stage I and II) rather than patients with regional or metastasized tumors (stage III and IV) (Fig. 2).
By Cox multivariate analysis, BRAF mutation was found to be an independent prognostic factor with the hazard ratio of 2.258 (p=0.029; 95% confidence interval, 1.08 to 4.69) (Table 4). Among the subtypes, mucosal type was shown to be a poor prognostic factor (p=0.001) as well as increased stage (p < 0.001), whereas female sex was seen to be a favorable prognostic factor (p=0.006) (Table 4).
5. Comparison of mutation status between primary melanoma and corresponding metastatic lesion and its influence on the outcome of melanoma patients
Among the 188 melanoma patients, 43 patients had primary melanoma and its corresponding metastatic lesions available for the study. Sixteen patients had lymph node metastasis only, 20 patients had distant metastasis only, and seven patients had both lymph node and distant metastases. Among the 43 patients, 55.8% (n=24) showed discordance in the mutational status between the primary and the corresponding metastatic lesion suggesting tumor heterogeneity (S2 Table). In uveal melanoma, 100% concordance was seen between the primary and corresponding metastatic tumors (Table 5, S3 Table).
The discordance between the paired tumors did not show significant association with age, sex, subtype, stage or type of metastasis (S2 Table). When the metastasis was found at the time of diagnosis of the primary tumor, it was designated as a “synchronous” metastasis, and when the metastasis was found after the initial diagnosis, it was designated as a “metachronous” metastasis. Thirty-one patients had metachronous metastasis, while 12 patients had synchronous metastasis. Among the 12 patients with synchronous metastasis, most of the metastasis was detected in the lymph nodes, except for the two patients whose metastases were observed in esophagus and brain. Among the 31 patients with metachronous metastasis, 15 patients did not receive any systemic therapy until the metastasis was found (The patient characteristics with metastatic tumors are listed in S3 and S4 Table).
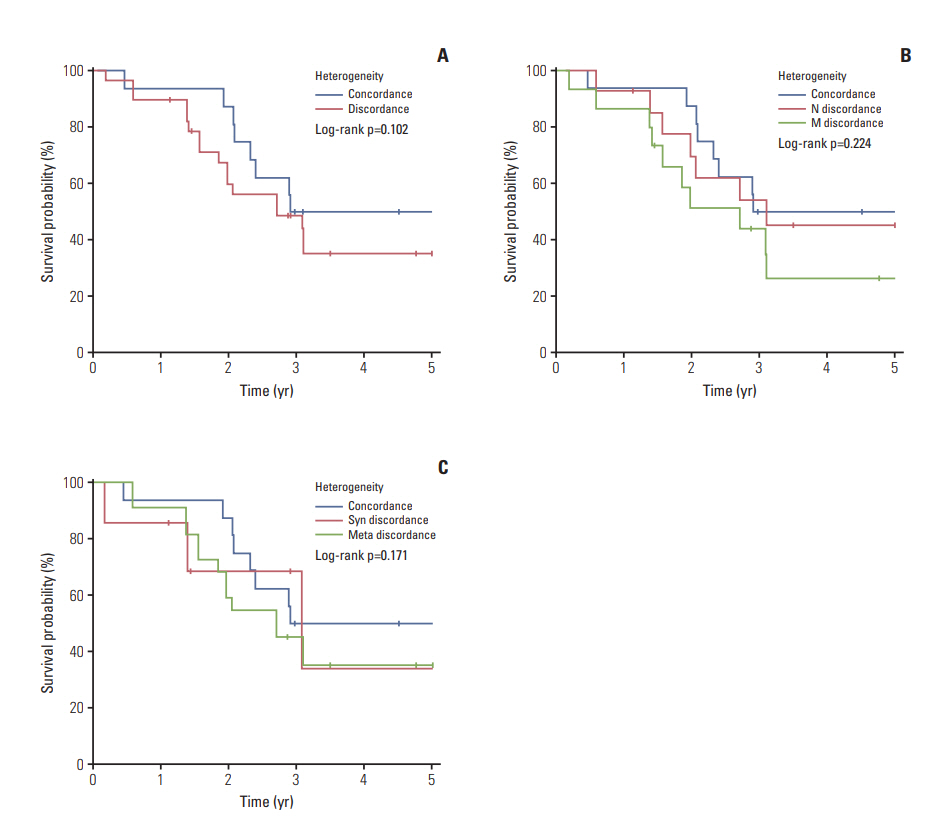
In the 31 metachronous metastatic tumors, the frequency of BRAF mutation and KIT amplification increased in metastatic tumors compared to its corresponding primary tumor. The increase of such genetic aberrations at metastatic lesion was similar in both lymph node metastasis and distant metastasis (Table 5). Although survival curve suggested poor survival rate in patient with genetic heterogeneity between primary tumor and metastatic tumor, especially distant metastasis, 5-year survival was not statistically different between the patient groups (Fig. 3).
Discussion
Metastatic melanoma is often challenging to treat as patients often exhibit drug resistance and experience tumor recurrence [11]. Resistance of tumors to drug therapy is at least partially explained by tumor heterogeneity, or the existence of multiple subclones of tumor cells with varying phenotype [12]. In melanoma, the concept of tumor heterogeneity was described as early as 1820 when a dissected melanoma metastasis was discovered to comprise of sections of different regions in primary melanoma [7]. Thus, this study aimed to identify the genetic heterogeneity within a set of well-known genetic alterations including BRAF V600, NRAS, and GNAQ/11 mutations, and KIT amplification in 188 Korean melanoma patients and its association with clinical features and impact on survival.
The overall incidence of somatic mutations within the BRAF V600 and NRAS genes were 17.6% and 12.6%, respectively, and KIT amplification was seen in 28.6%. GNAQ/11 mutation in the uveal type was found in 66.6%. Comparing with the Caucasian data, the frequency of NRAS mutation were less in our study especially in non-CSD type and CSD type (6.45% and 5.5%, respectively) [13]. This finding suggests the existence of ethnic difference in molecular pathogenesis of melanoma, even in same subtype. In addition, there was a pronounced discrepancy in the proportion of melanoma subtypes where the acral and mucosal types were more common compared to Caucasian data [14]. This finding is consistent with nationwide study investigating the incidence of melanoma in Korea [15].
Due to its aggressive progression, there has been continuous effort to find out the prognostic factors of melanoma [16]. Until now, the single most important prognostic factor for the survival in localized melanoma has been the tumor thickness [17]. Activating mutations of the BRAF oncogene have been reported in 33% to 47% of primary melanomas and 41% to 55% of metastatic melanoma [18]. The BRAFmutation constitutively activates BRAF and its downstream signaling of the mitogen-activated protein kinase pathway promoting proliferation, survival and spreading of tumor cells [19]. Although prognostic role of BRAF mutation in melanoma progression is still controversial [20,21], Long et al. [18] found that BRAFmutation associated with several features of highrisk melanoma, including earlier age of onset and shortened survival. In this study, although patients with BRAF mutation were in advanced stage at the time of diagnosis compared to BRAF wild-type patients, multivariate analysis revealed that BRAF mutation can also act as an independent prognostic factor, especially in the early stage melanomas (stage 1 and 2). Also, patients with BRAF mutation were younger compared to the BRAF wild-type patients which is consistent with previous reports [22,23].
During tumor progression, subclones frequently arise, which may result in differences in the proportion and pattern of specific aberrations between the primary and the metastatic or recurrent tumors that originate from it [24]. In our study, among the 43 patients with corresponding metastatic lesions, 55.8% of the patients showed discordance in the mutational status suggesting tumor heterogeneity. However, in our study, tumor heterogeneity did not show any association with clinical features. Although survival curve suggested negative effect of tumor heterogeneity on patient survival, there is no statistical difference in 5-year survival between the patients with genetic heterogeneity and homogeneity. Further study with a large sample size is necessary to conclude the effect of tumor heterogeneity on patient survival.
The frequency of BRAF mutation increased significantly in metastatic lesions compared to primary lesions. Our results are contrary to previous data indicating that BRAF mutations occur early in the development of melanoma and the incidence may not vary significantly during tumor progression [25,26]. However, sequential increase of BRAFmutation rate was reported in a subset of melanomas during progression of the disease [27-29]. Together, it is likely that BRAF mutations is not merely initial event in melanomagenesis but has a role in progression of melanoma.
In uveal melanoma patients, 100% concordance in mutation status between primary and metastatic tumors was observed. GNAQ and GNA11 mutations are known to occur in majority of uveal melanoma patients and are considered to occur early in uveal melanoma development [9]. Interestingly, several studies on uveal melanoma metastases describe that most metastases reflect the primary tumor [9] which may explain our results. However, further studies are required to clarify this prediction.
In conclusion, our study revealed that BRAF mutation was an independent prognostic factor of poor survival. Also, our data demonstrated tumor heterogeneity between primary melanomas and corresponding metastatic lesions for BRAF, NRAS mutations and KIT amplification, and genetic homogeneity in uveal melanoma for GNAQ/11. However, tumor heterogeneity did not show any significant association with clinical features and also had no significant impact on survival of the patients.