Introduction
Neuroblastoma is a childhood malignancy that arises from precursor cells of the sympathetic nervous system or the adrenal medulla. It is the most common extracranial solid tumor in children and accounts for 12% of all childhood cancer-related deaths [1]. The clinical course is highly heterogeneous, ranging from spontaneous regression without any therapeutic intervention to very fatal cases showing rapid progression and death despite modern intensive multimodal treatment. Because of these clinical heterogeneities, different therapeutic approaches are needed in the treatment of neuroblastoma. Traditionally, clinical features such as age at diagnosis and stage have been used to predict outcome and stratify risk groups in neuroblastoma. In addition to these clinical characteristics, many genetic and genomic features have been studied and incorporated into the modern prognostic classification of neuroblastoma [2].
Amplification of the MYCN oncogene was one of the first reported genetic markers for highly aggressive and advanced stage neuroblastoma. MYCN amplification is observed in approximately 20% of cases and remains a powerful prognostic factor indicating poor outcome [3]. Tumor cell DNA ploidy has also been shown to be independently correlated with outcome; near-triploidy and diploidy/tetraploidy are associated with excellent and poorer outcomes, respectively [4]. With the development of high-throughput techniques such as chromosomal or array comparative genomic hybridization, numerous recurrent large-scale genomic imbalances including loss of heterozygosity of chromosomal regions 1p, 3p, and 11q, along with gain of chromosome 1q and 17q, have been reported to be associated with poor outcome in neuroblastoma [5]. Recently, the International Neuroblastoma Risk Group project reported that segmental chromosomal alterations, rather than single genetic markers, have prognostic impact in neuroblastoma [6].
Activating mutations of the anaplastic lymphoma kinase (ALK) gene in neuroblastoma have been reported by several groups since 2008 [7-10]. ALK is a tyrosine receptor kinase which is encoded by the ALK gene located on the short arm of chromosome 2 (2p23.2). The oncogenic potential of ALK was first identified in anaplastic large cell lymphoma through the formation of a nucleophosmin-ALK fusion protein with constitutive kinase activity by chromosomal translocation t(2;5)(p23;q35). One of the first reports of ALK in neuroblastoma described the germline ALK mutation in the neuroblastoma pedigree. Mosse et al. [10] demonstrated that germline ALK mutations are responsible for susceptibility to hereditary neuroblastoma development. Subsequently, somatic ALK mutations have been reported in 6%-12% of sporadic neuroblastoma and these mutations are located mostly within the tyrosine kinase domain [8]. In addition to point mutations, ALK amplification has been reported in about 2%-6% of neuroblastoma cases by several studies [11,12].
Telomere maintenance is essential for cancer cell survival. There are two different mechanisms of telomere maintenance: one is through the increased expression of telomerases and the other is through the alternative lengthening of telomeres (ALT) pathway [13]. The former is related to telomerase reverse transcriptase (TERT) expression, and the latter is known to be associated with mutation of the alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene [13]. ATRX mutations have been found in neuroblastoma in older children and adolescents in particular. ATRX mutation was associated with loss of the nuclear ATRX protein, longer telomeres and neuroblastoma without MYCN amplification. This mutation was related to an indolent, but progressive course in neuroblastoma [14]. Recently, telomerase activation by genomic rearrangement in the region of proximal of the TERT gene was reported in neuroblastoma, and this rearrangement defined a subgroup of high-risk tumor with particularly poor outcome [15,16].
In this study, ALK mutation and amplification, ALK protein expression, loss of nuclear ATRX protein, and TERT protein expression were studied to investigate potential correlations between these molecular characteristics and clinical features or outcomes in neuroblastoma.
Materials and Methods
1. Patients and specimens
Patients who were histologically diagnosed with neuroblastoma and treated at Seoul National University Children’s Hospital from January 2002 to July 2012 were enrolled in this study. A list of patients was obtained from the hospital’s computerized database. A total of 104 patients were included initially, but 14 patients without initial tumor samples before chemotherapy were excluded. After a review of hematoxylin and eosin–stained slides, an additional five patients were excluded due to inadequate tumor specimens. A tissue microarray (TMA) was prepared with tumor samples from the remaining 86 patients, and a further 13 patients were excluded due to insufficient tumor specimens on the TMA slides. Finally, 72 patients were evaluated in this study.
Clinical data were retrospectively collected. This data included gender, age at diagnosis, tumor location, International Neuroblastoma Staging System (INSS) stage [1], risk group according to the Children’s Oncology Group risk stratification system [17], treatment history, clinical follow-up, and final outcome.
Formalin-fixed, paraffin-embedded (FFPE) tumor tissue obtained during surgery or biopsy was used. For patients with somatic mutations in their tumor tissue, peripheral blood was used to determine whether the mutations were somatic or germline.
This research was carried out in accordance with the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Seoul National University Hospital (H-1103-003-352). Informed consent was obtained according to the guidelines of our Institutional Review Board.
2. Immunohistochemical staining
The hematoxylin and eosin sections were reviewed by a pathologist, and the TMA was prepared from FFPE neuroblastoma tissue samples. Immunohistochemical (IHC) staining was performed on 4-μm-thick TMA sections using an ALK monoclonal antibody (Novocastra, Leica Biosystems, Newcastle upon Tyne, UK), a polyclonal antibody against ATRX (Sigma-Aldrich, St. Louis, MO) and a monoclonal mouse anti-TERT antibody (Novus Biologicals, Littleton, CO). The TMA sections were deparaffinized and rehydrated, and heat-induced epitope retrieval was performed by heating in Tris–ethylenediaminetetraacetic acid buffer, pH 9.0, at 100°C for 15 minutes. The intensity and area score of positive cells was evaluated and scored by a pathologist. The intensity score was defined as follows: 0, negative; 1, weak positive; 2, medium density positive; and 3, strong positive, and the area score was categorized as follows: 0, negative; 1, 1%-25%; 2, 26%-50%; 3, 51%-75%; and 4, > 76%. The intensity score was well correlated with the area score, so intensity score was used for data analysis. For ALK and TERT protein expression, a staining intensity score of 2 or more was defined as positive. Loss of the nuclear ATRX protein was defined as a staining intensity score of 0 or 1.
3. Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed on unstained TMA sections to investigate MYCN and ALK amplification. Probes used in this study included MYCN (2p24.1, orange; Vysis, Downers Grove, IL), centrosomal protein 2 (2p11.1-q11.1, green; Vysis) and an ALK Dual Color, Break Apart Rearrangement Probe (2p23; Vysis, Abbott Laboratories, Abbott Park, IL). Values for each signal and the ratios of red/green signals were reported in at least 50 non-overlapping nuclei per specimen. MYCN/ALK amplification was defined as over a 4-fold increase of the MYCN/ALK signal number relative to the number of control probe signals, in accordance with previous studies [2,18].
4. Mutation analysis
Genomic DNA was extracted from FFPE tumor tissue. Three to five 10-μm-thick sections were used for DNA extraction. For samples in which tumor content was below 70%, the tumor area was selectively scraped from the slides. Paraffin was removed from the FFPE sections using xylene followed by ethanol washes. The tissue was then lysed with lysis buffer and proteinase K overnight at 56°C. Lysates were incubated at 90°C for 15 minutes to remove DNA crosslinks, and genomic DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). For DNA extraction from peripheral blood lymphocytes, the QIAamp DNA Mini Kit (Qiagen) was used.
Polymerase chain reaction (PCR) amplification was performed for exons 23, 24, and 25 of the ALK gene. ALK PCR was performed in a 20 μL reaction mixture containing 5 μL of extracted template DNA, 2 μL of 10 pmol primers, and 10 μL of HotStarTaq Master Mix (Qiagen). PCR products were subjected to electrophoresis on 2% agarose gels and were purified using a QIAquick PCR Purification Kit (Qiagen). Direct sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on the ABI PRISM 3730XL Analyzer (Applied Biosystems). The primer sets used in this study are listed in Table 1.
5. Statistical analysis
Differences between categorical variables were measured by the chi-square test or Fisher exact test, and differences between means in continuous variables were calculated using Student’s t test. The Kaplan-Meier method and log-rank univariate comparisons were used to estimate survival. Cox proportional hazard regression model was used for multivariate analysis. Survival analysis was performed for all patients except for three patients who had not had a sufficient follow-up duration after their initial diagnosis or because of an absence of follow-up data. SPSS ver. 21.0 (IBM Corp., Armonk, NY) was used for all statistical analyses, and p < 0.05 was accepted as statistically significant.
Results
1. Clinical characteristics
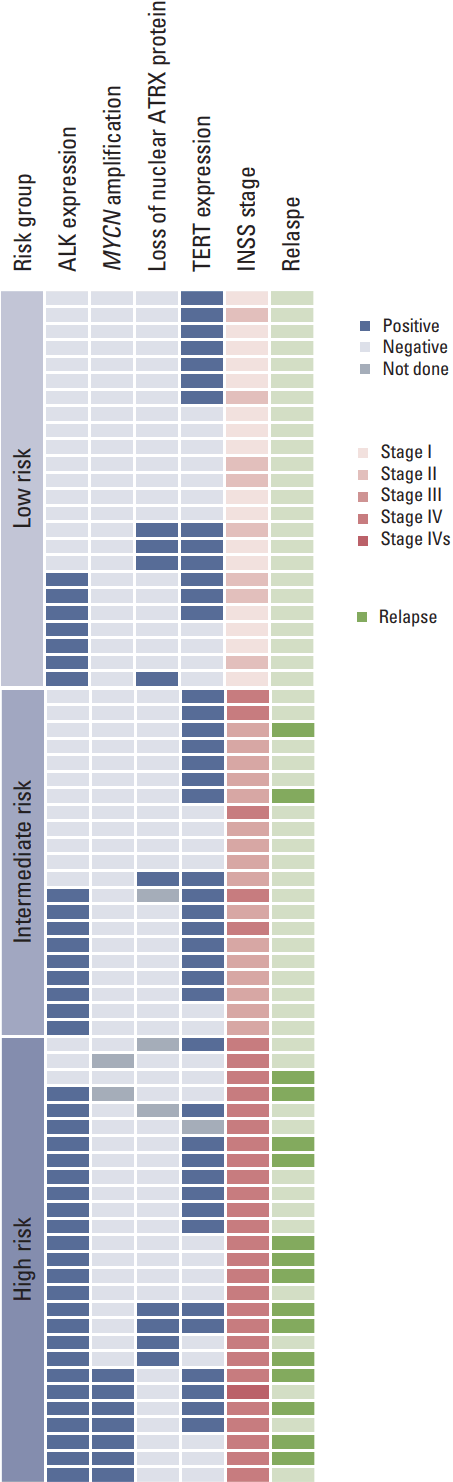
A total of 72 patients were enrolled in this study. The clinical characteristics of the patients are summarized in Table 2. The median patient age was 2.0 years (range, 0.0 to 16.7 years). The primary tumor sites were the retroperitoneum in 55 patients (76.4%), the posterior mediastinum in 15 patients (20.8%), and the neck in two patients (2.8%). MYCN amplification was evaluable in 70 tumor samples, and seven patients (10.0%) had MYCN amplification at initial diagnosis. Treatment was decided according to the patient’s age, stage, and MYCN status.
Sixteen patients relapsed (relapse rate, 29.3%), and treatment-related mortality occurred in three patients. The clinical and molecular characteristics of the relapsed patients are summarized in Table 3. In the 13 patients who died of disease, the median time to relapse from diagnosis was 23.6 months (range, 3.4 to 83.7 months), and the median time to death from relapse was 5.6 months (range, 1.2 to 55.3 months).
2. ALK protein expression
ALK protein expression was evaluable in 72 tumor samples, and samples from 40 patients (55.6%) showed ALK+ staining. The incidence of ALK+ staining increased in accordance with increasing tumor stage and risk group: 25.0% in stage I, 37.5% in stage II, 43.8% in stage III, and 80.6% in stage IV patients (p=0.001); 29.2% in low-risk, 42.9% in intermediate-risk, and 88.9% in high-risk (p < 0.001). There was no difference in patient age between ALK– and ALK+ patients (3.0±3.9 years vs. 3.1±3.2 years, p=0.88). Samples from all patients with MYCN amplification showed ALK+ staining by IHC. ALK+ staining was not associated with loss of nuclear ATRX protein or TERT protein expression.
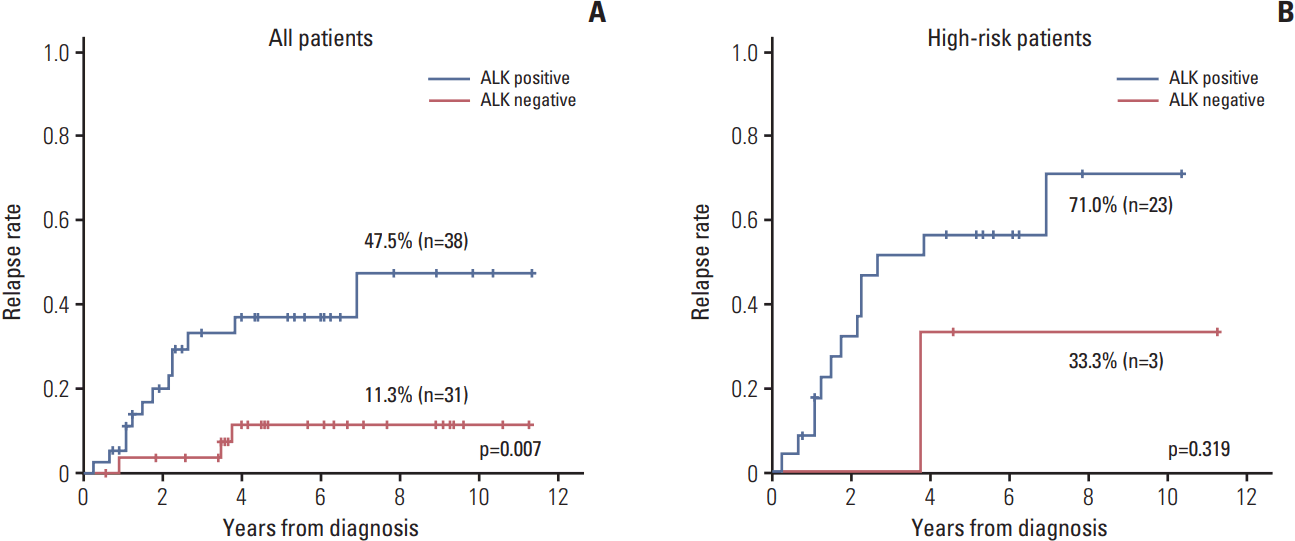
The relapse rate was significantly higher in ALK+ patients compared to that of other patients (47.5% vs. 11.3%, p=0.007) (Fig. 3A). However, most of the relapsed patients were high-risk patients, and only three patients in high-risk group showed ALK negativity. When the survival analysis was confined to the high-risk patients, there was no significant difference in relapse rate (Fig. 3B). In the multivariate analysis using ALK expression and risk group as variables, only the risk group retained the prognostic significance (p=0.003).
3. ALK amplification and mutation
ALK amplification was evaluable in 65 patients, but amplification was not observed in any of them. ALK mutation studies were carried out for 49 patients whose tumor samples were sufficient for DNA extraction. DNA sequencing of ALK revealed two missense mutations (F1174L and R1275Q) in two patients (Nos. 47 and 75). Sequence analyses of the constitutional DNA from peripheral blood showed that both were somatically acquired mutations.
One patient (No. 47) who carried a R1275Q mutation was a 4.6-year-old male who was diagnosed with INSS stage IV, MYCN–non-amplified neuroblastoma originating from the adrenal medulla with bone, bone marrow, and lymph node metastasis. ALK IHC results were positive. Even after induction chemotherapy, a significant decrease in tumor volume was not achieved, indicating stable disease. High dose chemotherapy and autologous peripheral blood stem cell transplantation (HDCT/aPBSCT) could not be administered because of persistent bone marrow involvement. The patient’s chemotherapy regimen was changed several times, but the disease progressed and the patient eventually died of disease 2.6 years after the initial diagnosis.
The patient (No. 75) with the F1174L ALK mutation was a 1.9-year-old girl who was diagnosed with poorly differentiated neuroblastoma with MYCN amplification. ALK+ staining was determined by IHC. The INSS stage was stage IV with bone, bone marrow and supraclavicular lymph node metastasis. After induction chemotherapy, tandem HDCT/aPBSCT was performed, followed by radiotherapy to the primary site and interleukin-2/isotretinoin treatment. The patient relapsed 3 months after the second HDCT/aPBSCT and died of disease which had rapidly progressed despite the salvage chemotherapy.
4. Loss of nuclear ATRX protein
ATRX expression was evaluable in 69 tumor samples. Nine patients (13.0%) showed loss of nuclear ATRX protein. The clinical and molecular characteristics of these nine patients are summarized in Table 4. MYCN amplification was not observed in any of them.
Among these nine patients, four patients had stage IV disease and belonged to high-risk group. All of these patients were older than 3 years of age; two were older than 10 years of years. Three of these patients relapsed, and the time to relapse from diagnosis were 25.9, 27.9, and 46.5 months respectively. Two of them (Nos. 23 and 38) survived for more than 4 years after relapse with an indolent disease course. Unexpectedly, five patients with stage I-III neuroblastoma (low- to intermediate-risk group) showed loss of nuclear ATRX protein, and all of them are alive to date without disease.
5. TERT protein expression
TERT protein expression was evaluable in 71 tumor samples, and samples from 42 patients (59.2%) showed TERT+ staining. The incidence of TERT expression did not differ according to tumor stage or risk group: 50% in stage I, 62.5% in stage II, 68.8% in stage III, and 56.7% in stage IV patients; 54.2% in low-risk, 71.4% in intermediate-risk, and 53.8% in high-risk patients. TERT+ patients were younger than other patients, but this was not statistically significant (2.2±2.7 years vs. 4.1±4.3 years, p=0.052). TERT expression was not associated with MYCN amplification, loss of nuclear ATRX protein or ALK protein expression.
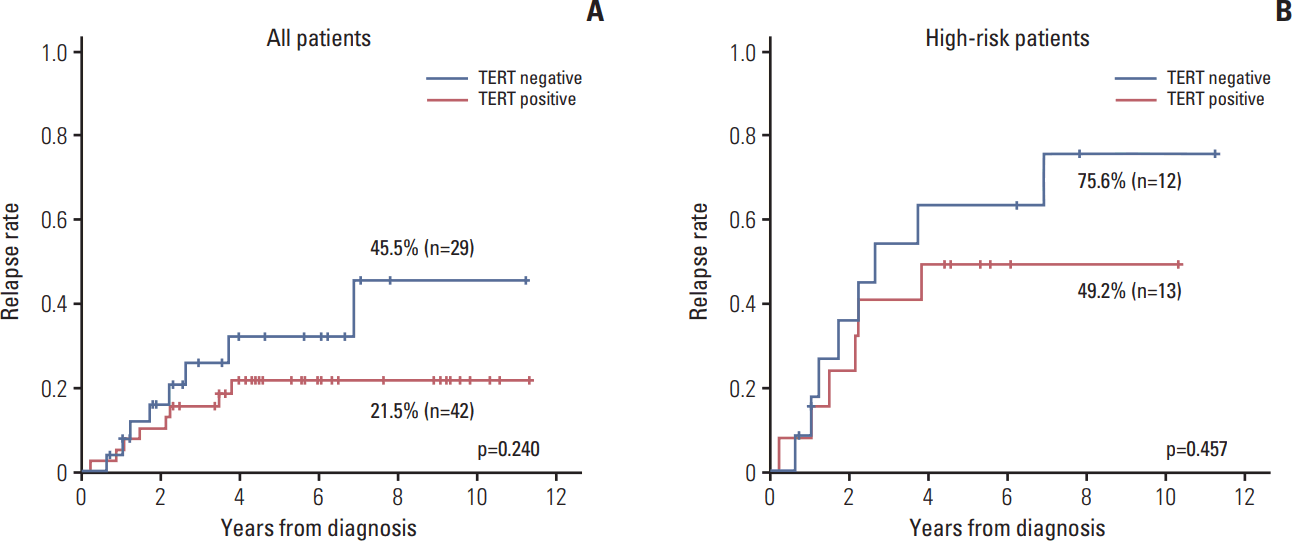
The relapse rate was slightly higher in TERT– patients compared with TERT+ patients, albeit without statistical significance (45.5% vs. 21.5%, p=0.240) (Fig. 4A). When the survival analysis was limited high-risk patients, the results were similar to the survival analysis for all patients (75.6% vs. 49.2%, p=0.457) (Fig. 4B).
Discussion
In this study, ALK mutation and amplification, ALK protein expression, loss of the nuclear ATRX protein, and TERT protein expression were studied to investigate potential correlations between these molecular characteristics and clinical features or outcomes in neuroblastoma. ALK mutation was found in a small population. No ALK amplification was observed in this group, but ALK expression was observed in a significant number of patients and ALK positivity was correlated with advanced stage and high-risk group. Loss of ATRX protein was observed in two different populations.
The oncogenic potential of ALK has been demonstrated by several in vitro and in vivo studies. Previous reports showed that neuroblastoma-derived cell lines harboring mutated ALK alleles exhibit constitutive ALK phosphorylation [9,10]. The F1174L and R1275Q variants possess the capacity to induce cytokine-independent growth in IL-3–dependent murine hematopoietic Ba/F3 cells, and two human neuroblastoma cell lines harboring the F1174L mutation have been shown to be sensitive to the inhibitor [8]. In a subsequent study, cell lines harboring F1174L-mutated ALK were relatively resistant to crizotinib showing differential inhibitor sensitivity [19], and this resistance was overcome by more potent inhibitor [20]. Taken together, these findings mean that the susceptibility to the inhibitor varies depending on the type of mutation and the drug.
ALK expression in neuroblastoma has also been investigated in a number of studies. Overexpression of ALK has been known in various cancers such as thyroid carcinoma, non-small cell lung cancer, breast cancer, melanoma, neuroblastoma, glioblastoma, astrocytoma, retinoblastoma, Ewing sarcoma and rhabdomyosarcoma [21]. In childhood cancer, there have been studies on ALK expression in rhabdomyosarcoma. ALK positivity in rhabdomyosarcoma was reported up to 45%, and it was related to the alveolar subtype, FOXO1 gene rearrangement, presence of metastasis and poor outcome [22,23]. Despite studies of ALK expression in various cancer, its mechanism in the pathogenesis of these cancers is unclear. Irrespective of the mechanism, the functional consequences of ALK expression has been investigated in several studies. Passoni et al. [24] observed aberrant ALK phosphorylation not only in a cell line carrying the R1275Q mutation but also in a high ALK-expressing cell line carrying a wild-type ALK gene. Furthermore, they found that a small molecule inhibitor targeting ALK exerted a potent cytotoxic effect on the high-ALK expressing wild-type cells [24]. In one study investigating transcriptomic characteristics, high-ALK expressing neuroblastoma showed similar global gene expression patterns to neuroblastoma with mutated ALK [25]. The molecular mechanism responsible for ALK overexpression needs further study including investigation of post-transcriptional modifications, but these findings suggest a positive correlation between ALK abundance and constitutive ALK activation.
The correlation between ALK IHC expression and clinical characteristics or treatment outcome in neuroblastoma has been evaluated in two previous studies. In one study by Passoni et al. [24], strong ALK protein expression was found up to 77% of advanced neuroblastoma, and it was significantly up-regulated in advanced/metastatic compared with localized neuroblastoma. In this study, overexpression of either mutated or wild-type ALK was defined as a poor prognosic factor, but clinical stage or risk group were not considered in the survival analysis. In the other studies by Duijkers et al. [26], ALK protein expression was more frequent in advanced stage neuroblastoma, but it was not related to the risk group showing ALK positivity in only 52% of the high-risk patients. In this study, higher percentages of ALK positive cells in patient samples correlated with inferior survival in univariate and multivariate analyses with established prognostic factors, such as stage, age, and MYCN status. In our present study, the incidence of ALK positivity was strongly correlated with both clinical stage and risk group. Even though there was a correlation between ALK positivity and poor survival in the analysis with all patients, but there was no prognostic value of ALK positivity when the survival analysis was confined to the high-risk patients. These findings suggest that ALK protein expression could be a marker related to the aggressive neuroblastoma, but it was not the independent prognostic factor for the outcome.
In this study, ALK mutations were found in two of 49 patients (4.1%), and ALK amplification was not observed. The incidence of ALK mutations was relatively lower compared to that in previous reports, but this can be partly attributed to the method of detection. In this study, only three exons harboring mutational hotspots were investigated using PCR and direct sequencing. On the other hand, ALK expression was observed in 40 patients (55.6%), and it was more frequently observed in advanced stage and high-risk neuroblastoma. Along with the previous studies, these findings suggest a possible role for ALK expression in the pathogenic mechanism of aggressive neuroblastoma, indicating the importance of elevated ALK expression irrespective of ALKmutation or amplification [24,25]. Relapsed or refractory neuroblastoma has a dismal prognosis and there is no effective treatment option to date. Molecular targeted therapy could be one option for this group of patients, but mutation at the ALK locus has been found in a limited number of cases, and the incidence of focal amplification is also very low. There are preclinical evidences that high ALK-expressing cell line exhibit ALK activation, and small molecule inhibitor exerted a cytotoxic effect on the high-ALK expressing wildtype cells [24]. Therefore, ALK-targeted therapy could be considered as one of the therapeutic strategy for neuroblastoma patients with an elevated level of expression of either wildtype or mutated ALK especially in the relapsed or refractory setting. The validity of this strategy needs to be confirmed by clinical study.
In this study, all of the patients with MYCN amplification were also positive for ALK expression, as determined by IHC. A correlation between ALK and MYCN in neuroblastoma has been investigated in a few studies. Schonherr et al. [27] showed that ALK activity was important in the initiation of MYCN transcription and that MYCN gene transcription was eliminated with the use of a specific ALK inhibitor. This ALK-induced transcription of MYCNwas found to be mediated by extracellular signal-regulated kinase 5 in a recently published study [28]. These findings support the results of this study that all of the MYCN-amplified neuroblastoma patients had ALK protein expression.
The association between telomere length and tumor prognosis has been studied in various types of cancer. In neuroblastoma, telomere length itself was studied as a significant prognostic parameter in a study showing that short telomeres were predictive of a favorable prognosis, whereas long or unchanged telomeres were predictive of poor outcome [29]. Recently, telomerase activation by genomic rearrangement in the region of proximal of the TERT gene was reported in neuroblastoma, and this rearrangement defined a subgroup of high-risk tumor with particularly poor outcome [15,16]. In these studies, TERT rearrangements were associated with increased TERT mRNA expression and upregulated enzymatic telomerase activity. In our study, TERT expression was assessed by IHC staining and TERT expression appeared to be inversely correlated with survival, but there was no statistically significance, and further study is needed to draw any conclusion.
A non-telomerase-dependent mechanism of telomere maintenance is ALT. A recent study has documented that ATRX is lost in 90% of in vitro immortalized cell lines with ALT [30]. ATRX acts as a suppressor of the ALT pathway, and mutation of ATRX is correlated with cancer cell survival through the ALT pathway. In the first report describing ATRX in neuroblastoma [14], ATRX mutations were found in 44% of tumors from adolescent and young adult patients with metastatic neuroblastoma, and in none of the tumors from infants with metastatic neuroblastoma. The children whose tumors had ATRX mutations were typically older than 5 years of age or had a chronic or indolent course of disease. The ATRX mutations were mutually exclusive of MYCN amplification, and associated with loss of the nuclear ATRX protein, longer telomeres, and ALT [14]. In our study, ATRX expression was assessed by IHC staining. Unlike the previous report that evaluated ATRX in only metastatic neuroblastoma [14], ATRX expression was studied in all of the patients in this study. Nine patients (13.0%) displayed loss of nuclear ATRX protein, and none of them had MYCN amplification, similar to the previous study. Among the four stage IV patients, two patients were older than 10 years of age at diagnosis, and showed a very indolent disease course with late relapse and survival for longer than 4 years after the relapse. Interestingly, loss of ATRX protein was found not only in stage IV patients, but also in five patients with stage I-III neuroblastoma. These lower-stage patients were younger than the stage IV patients, and the youngest patient was 0.2 years of age. This finding suggests that there may be two different populations with loss of ATRX protein: (1) older patients showing an indolent disease course, and (2) young children with lower stage with a better prognosis. Additional research is needed on the meaning of the loss of ATRX protein in young patients.
One thing to consider in our study is that TMA slides were used for IHC and FISH studies. TMA preparation is a convenient method for various molecular studies, but it may not exactly reflect the nature of the actual tumor. To minimize this effect, the pathologist reviewed the original slide and produced a TMA slide as a representative part. One of the other limitations is that conventional PCR and Sanger sequencing were used for mutation analysis, and only three exons were examined. Considering the tumor heterogeneity and the sensitivity of the test, the mutation frequency could be actually higher than the results of this study when the ALK gene is examined using the next generation sequencing technique.
In summary, although ALK mutation was rare and no amplification was observed, ALK protein expression was found in a significant number of patients and was correlated with advanced stage and high-risk neuroblastoma. ALK protein expression could be considered as a marker related to the aggressive neuroblastoma, but it was not the independent prognostic factor for the outcome. Loss of ATRX protein was observed in two different populations, and further study is required to determine the meaning of ATRX loss in neuroblastoma.