Introduction
Cell death is crucial for normal development, homeostasis, and the prevention of hyperproliferative diseases such as cancer. Historically cell death has been categorized based on morphologic observations. Three major forms of cell death are recognized to date: apoptosis (type I cell death), autophagy (type II), and necrosis (type III or accidental cell death) [1]. Besides, there are alternative cell death pathways, such as anoikis, pyroptosis, cornification [2]. It is once thought that almost all regulated cell death (RCD) in mammalian cells resulted from apoptosis, but this view has been challenged by the discovery of several regulated nonapoptotic cell death pathways, one of them is ferroptosis [3-5].
Ferroptosis is generally referred as a mode of RCD involving the production of iron-dependent reactive oxygen species (ROS), which is distinct from other forms of cell death based on morphological, biochemical, and genetic criteria [5-7]. Ferroptotic cells exhibit completely different changes under transmission electron microscopy: smaller mitochondria with increased membrane density and reduced cristae [2,5,6]. Besides, inhibition of apoptosis, necrosis, and autophagy by small molecule inhibitors cannot reverse ferroptosis [5]. Mechanistically, iron metabolism and lipid peroxidation signaling are increasingly recognized as central mediators of ferroptosis, and multiple molecules affecting accumulation of lipid peroxidation products and lethal ROS derived from iron metabolism have been identified as inducers or inhibitors of ferroptosis. Common inducers for ferroptosis are erastin (inhibit cystine/glutamate antiporter system Xc−), RSL3 (inhibit glutathione peroxidase [GPX] 4), buthioninesulphoximine (inhibit glutamate-cysteine ligase), and common inhibitors are ferrostatin-1 (inhibit ROS), deferoxamine (inhibit fenton reaction), β-mercaptoethanol (enhance cystine/glutamate antiporter system Xc−), ciclopirox (intracellular iron chelator) [5-11].
At present, apoptosis evasion, also enhancement of antiapoptosis ability, is considered as the major reason for tumor refractory; as a mode of RCD completely independent from apoptosis, ferroptosis may provide a new therapeutic approach to cancer treatment. Recently, sorafenib, a targeted therapy that blocks cellular oncogenic kinases, has been proved to exert its cytotoxic effects on hepatocellular carcinoma cells through ferroptosis [12,13]. Considering this fact, we are wondering whether this new way of cell death exists in classical therapeutic drugs such as cisplatin, fluorouracil (5-FU), etc. Indeed, activation of an alternative cell death pathway may open up a new way for the utility of chemotherapeutic agents and overcome the related drug resistance.
In the present study, a number of classic chemotherapeutic drugs were screened for the occurrence of ferroptosis, and cisplatin was considered as an inducer for both ferroptosis and apoptosis in A549 and HCT116 cells. With further exploration, we highlighted that the depletion of reduced glutathione (GSH) and inactivation of GPXs played the vital role in the whole process of cisplatin induced ferroptosis. Besides, combination therapy of cisplatin and erastin were executed on A549/HCT116 cells, and the results demonstrated a significant synergistic effect on their anti-tumor activity. All these findings indicated that ferroptosis has a high potential to be a new opportunity for therapeutic intervention on cancer treatment.
Materials and Methods
1. Cell lines
Non-small cell lung cancer (NSCLC) cell lines A549, NCIH358, NCIH460, and Calu-1, human colorectal cancer cell line HCT116, human fibrosarcoma cell line HT-1080 were obtained from China Center for Type Culture Collection (CCTCC, Wuhan University, Wuhan, China). Cells were cultured in RPMI-1640 medium (Hyclone, Logan, UT) with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY), supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Hyclone). Cells were maintained in a humidified incubator at 37°C, in the presence of 5% CO2. All cell lines were cultured to limited passage before implantation and were routinely screened to confirm the absence of contamination.
2. Preparation of the reagents
Cells were under the exposure to several kinds of reagents. The amount and conditions of those reagents were as follows: cisplatin (Sigma, St. Louis, MO) was dissolved in normal saline (NS) with a final working concentration of 5 μg/mL, 5-FU (Sigma) was dissolved in NS with a final working concentration of 20 μg/mL, adriamycin (Meilunbio, Dalian, China) was prepared in dimethyl sulfoxide (DMSO; Sigma) with a final working concentration of 10 μg/mL, paclitaxel (Meilunbio) was prepared in DMSO with a final working concentration of 10 μmol/L, and sulfasalazine (Sigma) was prepared in DMSO with a final working concentration of 0.8 mmol/L. Erastin (Selleck, Houston, TX) was prepared in DMSO with a final working concentration of 10 μmol/L, ferrostatin-1 (Sigma) was prepared in DMSO with a final working concentration of 0.5 μmol/L, deferoxamine (Sigma) was prepared in DMSO with a final working concentration of 50 μmol/L, a stock solution of β-mercaptoethanol (Sigma) was diluted into a final working concentration of 50 μmol/L, a stock solution of z-vad-fmk (Beyotime, Haimen, China) was diluted into a final working concentration of 20 μmol/L, necrostatin-1 (Santa Cruz Biotechnology, Santa Cruz, CA) was prepared in DMSO with a final working concentration of 30 μmol/L, chloroquine (Meilunbio) was dissolved in NS with a final working concentration of 5 μg/mL.
3. Determination of cell viability
Cells (5×103) cultured in RPMI-1640 medium with 10% FBS were seeded in 96-well plates and received treatments for a given period. The cytotoxic effect of different treatments was determined by the methylthiazoltetrazlium (MTT; Biosharp, Carlsbad, CA) dye uptake method. MTT solution (20 μL, 5 mg/mL) was added to each well. After incubation for 4 hours at 37°C, the supernatants were removed and 150 μL DMSO were added to each well, then incubated at 37°C for 15 minutes. Optical density (OD) was detected with a microplate reader (Biotech, New York, NY). Inhibition rate (IR) (%)=[1–(OD of the experimental sample/OD of the control)]×100%, Q=IRA+B/(IRA+IRB–IRA×IRB) (IRA was the IR of drug A, IRB was the IR of drug B, IRA+B was the IR of drug A+drug B), and Q > 1.15 was a symbol for significant synergistic effect of drug A and drug B.
4. Determination of ROS levels
The 2×105 of cells were seeded in 6-well plates in advance. The next day, cells were incubated in 2 mL media with different drugs for 48 hours. Then, the culture media was replaced by serum-free media containing 10 μmol/L 2',7'-dichlorodihydrofluorescein diacetate (Sigma) and placed in dark for 30 minutes, gently shaken every 5 minutes. Cells were harvested in 15 mL tubes by centrifuging at 1,000 rpm for 5 minutes and washed 3 times with serum-free media followed by re-suspending in serum-free media, then incubated with 5 μL 7-aminoactinomycin D (KeyGEN Bio-TECH, Nanjing, China) in dark for 5 minutes. Fluorescence was determined by flow cytometry (BD Biosciences, San Jose, CA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. The average intensity of fluorescence in each group indicated the amount of ROS within cells.
5. Determination of reduced GSH levels
The 2×105 of cells were seeded in 6-well plates in advance. The next day, cells received different treatment for 48 hours followed by harvesting to determine cell number. Nearly 6×104 live cells from each sample were transferred to new tubes, washed in phosphate buffered saline (PBS) and centrifuged at 1,200 rpm at 4°C for 5 minutes twice. The cell pellet was resuspended in 8 μL protein removal solution, thoroughly incorporated, and placed in –70°C and 37°C sequentially for fast freezing and thawing, then placed in 4°C for 5 minutes and centrifuged at 10,000 ×g for 10 minutes. The supernatant was used to determine the amount of GSH in the sample. We used the GSH and GSSG Assay Kit (product No. S0053, Beyotime) and followed the product instructions to determine GSH levels [10]. Briefly, GSH assay buffer, GSH reductase, 5,5′-dithio-bis 2-nitrobenzoic acid solution and supernatant sample were mixed together and incubated at 25°C for 5 minutes, then NADPH was added into this system to trigger the reaction. The increase in the absorbance of 5-thio-2-nitrobenzoic acid was measured at 412 nm, and the GSH levels were calculated following the product instructions.
6. Determination of GPXs
Cells with or without compound treatment were harvested and washed with PBS twice, resuspended in 50 μL cell lysis solution and homogenized at 4°C for 15 minutes. The resulting crude lysates were cleared by centrifuging at 12,000 ×g for 10 minutes, and the supernatant was kept for next step. The total amount of protein in the supernatant was determined first through BCA method (BCA Protein Detection Kit, Google Bio, China). Then, Total Glutathione Peroxidase Assay Kit (product No. S0056, Beyotime) was used to measure the activity of GPX [10]. In microtubes, GPX assay buffer, NADPH, GSH, GSH reductase and supernatant sample were transferred and completely mixed according to the manufacturer’s guidelines. GPX reaction was started by adding peroxide reagent tert-butylhydroperoxide (t-Bu-OOH) to the mixture above. The decrease in the absorbance of NADPH was determined at 340 nm. The amount of enzyme which oxidizes 1 μmol of NADPH per min at 25°C is defined as one unit of enzyme.
7. Transmission electron microscopy
Cells were examined at the ultrastructural level using transmission electron microscopy (Olympus, Tokyo, Japan). At least three independent fields were acquired for each experimental condition. Representative photographs from one field of view are shown.
8. RNA Interference
Small interfering RNAs (siRNA) targeting IREB2 (siRNA IREB2-7499, GCUGUGAAAUUGUUUCGAAtt; siRNA IREB2-7500, GGAACAUUUUCUUCGCAGAtt) and siNeg (random sequence) were obtained from Genepharma Technologies (Shanghai, China). Six thousand cells were seeded in 96-well plates and incubated at 37°C for 24 hours. Reverse transfection was performed by preparing two solutions of siRNA-Optimem (siRNA:Optimem=1:100) and lipo2000-Optimem (lipo2000:Optimem=1:100, Lipofectamine 2000 and optimum media were both purchased from Invitrogen, Carlsbad, CA). After stayed at room temperature for 5 minutes, the two solutions were mixed up at a ratio of 1:1 and placed for 25 minutes at 37°C. Next, 50 μL siRNA-lipo2000-Optimem mixture and 50 μL serum-containing media were added to each well and incubated for 6 hours at 37°C. Following incubation, 100 μL serum-containing media were transferred to each well and incubated at 37°C. Real-time polymerase chain reaction and western blot were performed 48 hours later to verify the efficiency of transfection. Then, drug treatment experiments were performed 24 hours after the transfection.
9. Real-time polymerase chain reaction
Cells received siRNA transfection were harvested and RNA was purified using the Total RNA extraction Trizol kits (Invitrogen) according to the manufacturer’s instructions, the concentration of RNA was determined by spectrophotometer (Eppendorf, Hamburg, Germany). The 800 ng total RNA per sample was subsequently used in a reverse transcription reaction using the Reverse Transcription Kit (Takara, Tokyo, Japan). The primers for polymerase chain reaction (PCR) were designed with Primer Express (upstream sequence, 5′-CTACCTGCCGAGGATCTTGTGA-3′downstream sequence, 5′-TGATGAGCCATTCCAGTTCCAG-3′).
Quantitative PCR was performed on triplicate samples, and the relative amount of the target gene in experimental and control conditions was computed using the 2-ΔΔCT method.
10. Western blot
Cells received siRNA transfection were incubated with 50 μL cell lysis solution for 30 minutes, then transferred into new tubes and centrifuged at 16,000 rpm at 4°C for 15 minutes. The amount of protein in the supernatant was determined by BCA method (BCA Protein Detection Kit, Google Bio). After quantification, samples were mixed with sodium dodecyl sulfate (SDS) loading buffer and separated by SDS-polyacrylamide gel electrophoresis (Western Blotting Kit, Google Bio). Western transfer was performed using the iBlot system (Invitrogen). PVDF membranes (Millipore, Billerica, MA) were blocked for 1 hour in Tris-buffered saline (pH 7.4) with 1% Tween-20 (TBS-T) with 5% milk and incubated in primary antibody overnight at 4°C. Following washed about 10 minutes in TBS-T for three times, the membrane was incubated with secondary antibodies for 1 hour at 37°C. The membrane was washed again in TBS-T thrice for 10 minutes prior to visualization using enhanced chemiluminescence (ECL, Thermo Fisher Scientific, Waltham, MA). Antibody for IREB2 (ab10-6926, Abcam, Cambridge, MA) were used at 1:1,000 dilution, and detected using a goat mouse horseradish peroxidase conjugate secondary antibody (Santa Cruz Biotechnology) at 1:5,000 dilution.
Results
1. Cisplatin induced both ferroptosis and apoptosis in A549 and HCT116 cells
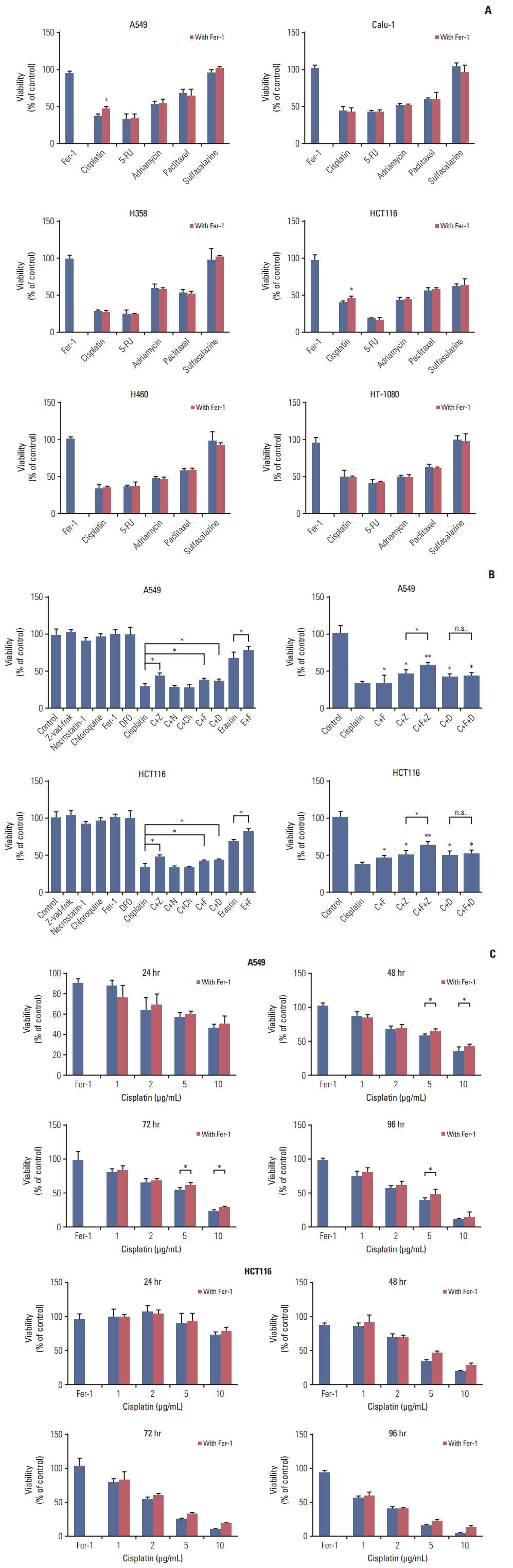
To test our hypothesis that ferroptosis might exist in the process of cell death induced by classical therapeutic drugs, we designed our exploration using six kinds of cancer cells and five typical chemotherapeutic drugs. Ferrostatin-1 has been proved to be a potent specific inhibitor of ferroptosis, and its mechanism of action is to prevent ferroptosis-induced accumulation of cytosolic and lipid ROS [5]. NSCLC cell lines A549, NCIH358, NCIH460, Calu-1, human colorectal cancer cell line HCT116, and human fibrosarcoma cell line HT-1080 received treatments of cisplatin, 5-FU, adriamycin, paclitaxel, and sulfasalazine for 48 hours, respectively. We found that only cisplatin induced cell death could be partially reversed by ferrostatin-1 in both A549 and HCT116 cells, while other drugs showed no response to ferrostatin-1 (Fig. 1A). Later, we applied several different kinds of cell death inhibitors together with cisplatin to A549 and HCT116 cells, including caspase inhibitor z-vad-fmk, necrosis inhibitor necrostatin-1, autophagy inhibitor chloroquine, ferroptosis specific inhibitor ferrostatin-1 and iron chelator deferoxamine. The results demonstrated that the cell death induced by cisplatin could be partially reversed by deferoxamine, ferrostatin-1, z-vad-fmk, and the phenomena became more obvious when ferrostatin-1 and z-vad-fmk were combined together (Fig. 1B). Thus, we thought that cisplatin induced cell death was a result from the joint action of both ferroptosis and apoptosis. Besides, we investigated the appropriate concentration and action time for cisplatin and found out that ferroptosis were more obvious when the concentration of cisplatin was higher than 5 μg/mL and the treatment was longer than 48 hours (Fig. 1C).
2. Cisplatin induced ferroptosis with proofs from multiple aspects
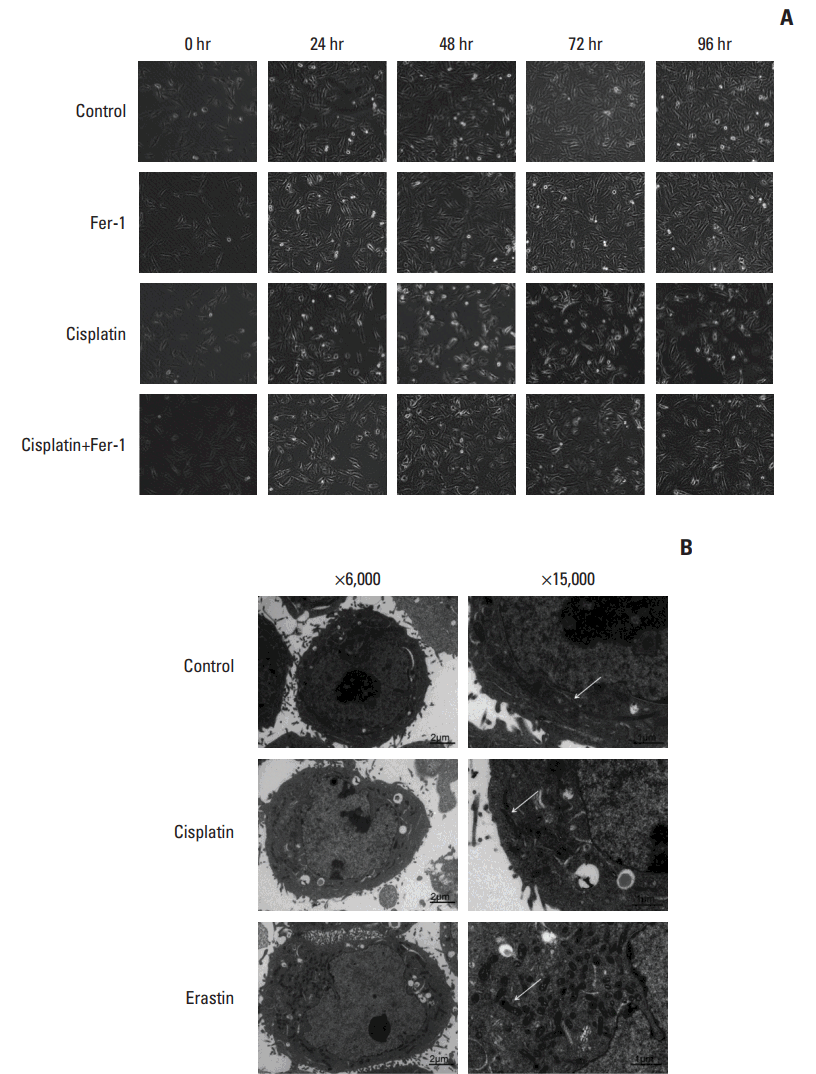
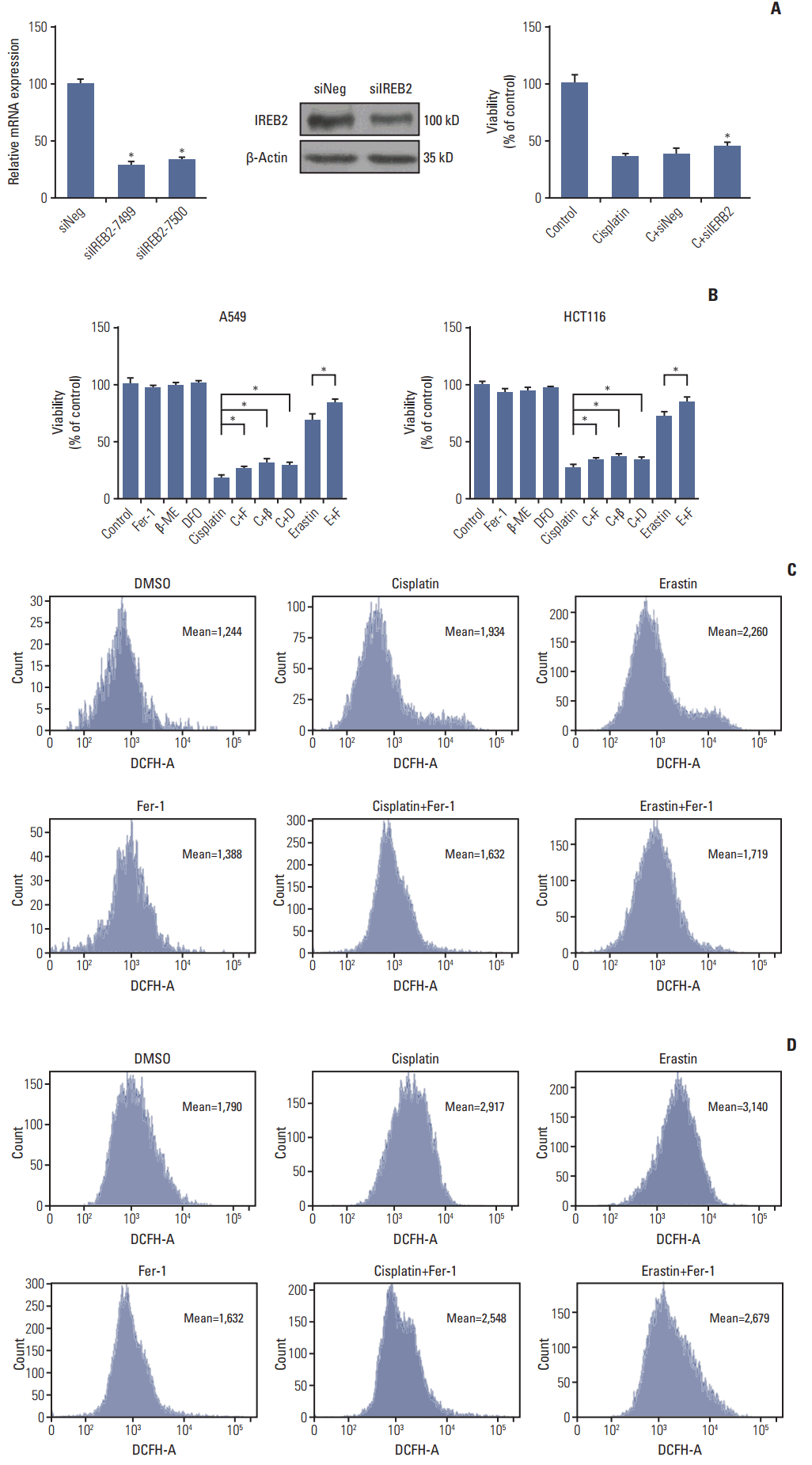
We further confirmed the occurrence of ferroptosis induced by cisplatin through multiple aspects. Firstly, optical microscopy images showed the protection of ferrostatin-1 from cisplatin in A549 cells (Fig. 2A). Secondly, electronic structural changes were observed. Mitochondrial changes are usually observed in ferroptosis and considered as the primary distinguishing feature [6]. Consistent with observations from previous reports, an alternation in mitochondrial ultrastructure was observed in both cisplatin and erastin (using as a positive control) treated HCT116 cells: mitochondria appeared on average smaller, less tubular, darker-staining membranes with distinct disrupted inner membrane foldings (Fig. 2B). Thirdly, the necessity of iron was determined. Ferroptosis is defined as a kind of iron-dependent cell death, so the involvement of iron is a key feature in this process. Iron-responsive element binding protein 2 (IREB2) is a master regulator of iron metabolism; silencing of IREB2 results in reciprocal changes in the iron uptake, mechanism, storage, and the inhibition of erastin induced ferroptosis [5,14,15]. We established siRNA-IREB2 (siIERB2) and transfected it into HCT116 cells. Results demonstrated that silencing IREB2 partially reversed the cytotoxicity of cisplatin, indicating the involvement of iron in cisplatin induced cell death (Fig. 3A). Fourthly, different kinds of ferroptosis specific inhibitors were applied. As displayed in our results, ferrostatin-1, deferoxamine and β-mercaptoethanol all showed suppressive effects on the anti-tumor effect of cisplatin (Fig. 3B). Fifthly, ROS levels were examined through flow cytometry analysis. Cells exposed to cisplatin displayed an obvious increase in ROS levels, and this phenomenon was partially reversed by ferrostatin-1 (Fig. 3C). Taken together, we concluded that ferroptosis was indeed involved in the process of cisplatin induced cell death, and we expected this new way of cell death could open up a new way for the utility of cisplatin in clinic.
3. GSH-GPXs system involved in the underlying mechanism of cisplatin induced ferroptosis
According to previous researches on ferroptosis, biomolecules like system Xc−, GSH, GPXs and intracellular iron are referred as key targets for ferroptosis regulators [16]. Erastin is determined to inhibit the function of system Xc−, leading to the depletion of GSH and inactivation of GPX4, and finally triggers ferroptosis [5,10,16]. Platinum compounds are proved to have a high affinity to thiol-rich biomolecules, and GSH is one of the most abundant non-protein thiols in cells; in cytoplasm, a major fraction (~60%) of the intra-cellular cisplatin is conjugated with GSH to form the Pt-GS complex [17,18]. Taking consideration of this feature, we supposed that GSH and GPXs were the most possible targets for cisplatin.
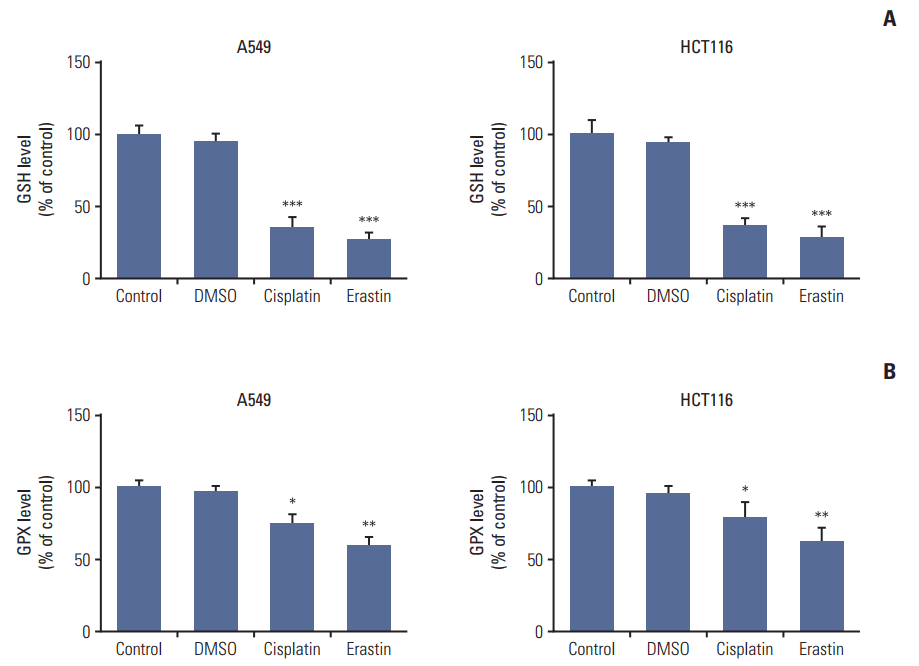
There, we treated A549 and HCT116 cells with either erastin (using as a positive control) or cisplatin, and collected cells to detect changes in GSH level and GPXs activity. Both erastin and cisplatin significantly decreased GSH levels and GPXs activities in the two kinds of cells; however, the inhibition of GPXs in cells treated with cisplatin was not as strong as those treated with erastin (Fig. 4). Previous data [10,16] suggested that GPXs activity owned a more vital role in the process of ferroptosis than GSH depletion, thus the weaker inhibition of GPXs induced by cisplatin defined it as a less potent inducer of ferroptosis.
It now appears that the ability of cisplatin to trigger ferroptosis is determined mainly by direct depletion of intracellular GSH. There are eight isoforms of GPXs in humans with different tissue expression and substrate specificities, and GPX4 is proved to be a central regulator of ferroptosis induced by erastin, which one plays the vital role in cisplatin induced ferroptosis worth further discussion.
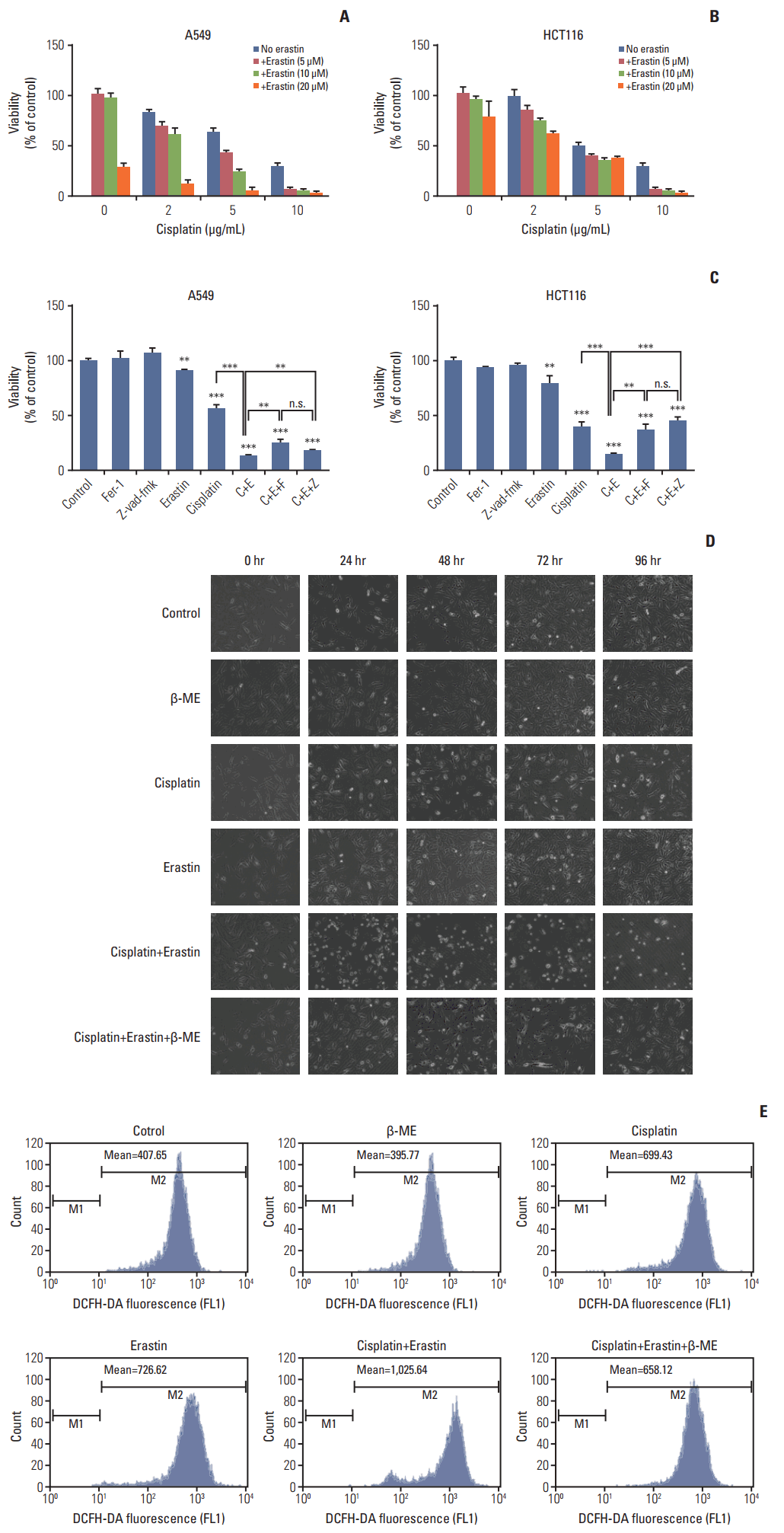
4. Additive effect observed in combination therapy of cisplatin and erastin
After verifying the occurrence of ferroptosis in cisplatin induced cell death, we investigated whether ferroptosis was able to become a potent assistance to cisplatin’s lethality. A549 and HCT116 cells were exposed to cisplatin alone or cisplatin together with erastin in different concentrations for 48 hours, and we found that the toxicity to cells improved a lot when the two drugs were combined together (Fig. 5A and B). The combination coefficients of cisplatin and erastin were always more than 1.15 when both drugs were in lower concentrations, which meant significant synergistic effect (Tables 1 and 2). In the subsequent experiments, we found that this improvement was significantly blocked by ferroptosis specific inhibitor ferrostatin-1, β-mercaptoethanol, and caspase inhibitor z-vadfmk in A549 and HCT116 cells, indicating the joint action of apoptosis and ferroptosis (Fig. 5C and D). Besides, elevated ROS level was observed in combination therapy and it could be significantly reversed by β-mercaptoethanol (Fig. 5E). Our data suggested that cisplatin together with erastin demonstrated better anti-tumor activity than any of them alone. Considering the completely different mechanism of ferroptosis, combining erastin together with cisplatin could be a good strategy to enhance the utility of cisplatin.
Discussion
Ferroptotic death is morphologically, biochemically, and genetically distinct from apoptosis, various forms of necrosis, and autophagy [5]. The present work was initiated as an attempt to understand whether this regulated nonapoptotic cell death pathway could be an alternative cell death mechanism for those classic chemotherapeutic agents. Our observations, for the first time, demonstrated that cisplatin was an inducer for both ferroptosis and apoptosis in A549 and HCT116 cells, and the GSH depletion together with the inactivation of GPXs played a vital role in the underlying mechanism. Furthermore, combination therapy of cisplatin and erastin showed significant improvement on their antitumor activity.
Among various anti-cancer agents, cisplatin is one of the most effective and widely used anti-cancer agents for the treatment of solid tumors [19]. The cytotoxic effects of cisplatin are thought to be mediated primarily by the generation of nuclear DNA adducts, causing apoptosis [20]. Unfortunately, many cancers initially responds to cisplatin but then the tumor come back and are frequently refractory to further platinum therapy [21]. Resistance to cisplatin is concluded occurring by the following molecular mechanisms: altered cellular accumulation of drug, altered DNA repair and cytosolic inactivation of drug [22,23]. These mechanisms all affect on the cisplatin-induced apoptosis but don’t influence cisplatin-induced ferroptosis. Herein, ferroptosis, as a new mode of regulated cell death completely independent from apoptosis, might open up a new way to solve the problem of tumor resistance to cisplatin. According to our study, cisplatin combined ferroptosis inducer erastin together showed significant additive effect on their anti-tumor activities, which was consistent with the results from Roh et al. [24]. Both ferroptosis and apoptosis attributed to this improvement. Thus, ferroptosis provided us with a chance to change the old impression on cisplatin, and brought new thoughts for the utility of cisplatin in clinic, even new strategies to solve the problem of tumor resistance.
Not all cancer cells are sensitive to ferroptosis inducers; in order to get better use of this new way of cell death, investigations are carried out to find out what kind of cells are more sensitive to ferroptosis. Some relationships between ferroptosis and cells’ genetic background are revealed, and RAS is the most famous among all the reported genes [16]. Yang and Stockwell [7] revealed that Ras mutation contributed to the iron overload in cells and made them more sensitive to ferroptosis inducers. In clinic, activating mutations in the Ras oncogenes are found in ~20% of all human tumors and always associated with drug resistance, defining a subset of patients for whom prognosis is poor and treatment options are limited [25-28]. Thus, ferroptosis is regarded as a promising pharmacological target for Ras-mutant tumors.
However, sensitive cancer subtypes are still uncertain. Ras mutation is not specific enough to be a symbol for ferroptosis. According to the profiles of erastin sensitivity for across a panel of 117 cancer cell lines, cell lines with Ras mutation are on average no more sensitive to ferroptosis-inducing compounds than wild type cells [10]. This was also reflected in our study. A549, HCT116, and Calu-1 used in our experiments were all K-Ras mutation cell lines, but ferroptosis was only observed in A549 and HCT116 cells.
Besides, there was another controversy. In our study, we didn’t observe ferroptosis in sulfasalazine treated tumor cells, while Gout et al. [9] found sulfasalazine to be a potent inhibitor for the cystine/glutamate antiporter system Xc− and Dixon et al. [5] demonstrated it to be an inducer for ferroptosis with lower potency than erastin. There were several possible reasons: different experiment conditions like the drug concentration and action time; or the method we used was not sensitive enough to detect faint ferroptosis induced by sulfasalazine; or the cell lines we chose were not as sensitive to sulfasalazine as lymphoma cell lines.
Ferroptosis is a newly discovered mode of cell death, although we are full of expectations for it, many questions are waiting to be answered, more specific genotype symbols or biomarkers for the occurence of ferroptosis, suitable drugs and their usage, and other combination therapies should be explored. There is still a long way to go before it eventually applied to clinic.
In conclusion, the present study, for the first time, determined that cisplatin was an inducer for both ferroptosis and apoptosis in A549 and HCT116 cells, and the GSH depletion together with the inactivation of GPXs played vital roles in the underlying mechanism. Further investigations showed that combining cisplatin together with erastin gained a significant synergistic effect on their anti-tumor activity. Thus, our results demonstrated that ferroptosis had a great potential of being a new approach in anti-tumor therapies and opened up a new way for the utility of classic drugs.