Introduction
Genomic alterations are a major cause of cancer initiation and progression [1,2]; however, they do not account for all of the changes in gene expression that are observed in cancer. RNA editing is a post-transcriptional molecular modification process that changes specific nucleotide sequences within an mRNA molecule after it has been generated by RNA polymerase. Therefore, RNA editing contributes to the diversity and plasticity of cellular RNA signatures along with amino acid changes, alternative splicing, and regulation by small RNA species [3].
In mammals, two classes of RNA editing have been well characterized, cytidine to uridine (C-to-U) and adenosine to inosine (A-to-I) [3,4]. In humans, the most common type of RNA editing is A to I, which is catalyzed by the adenosine deaminase acting on the RNA (ADAR) enzyme. Adenosine is catalyzed by adenosine deaminase, then converted to inosine. Inosine base pairs with cytosine and is read as guanine by reverse transcription and translation machinery [5].
The recent development of next generation sequencing technologies has caused the number of identified RNA edits to increase dramatically. Several groups have been studying new methods to detect RNA editing [6,7], leading to identification of several million potential RNA editing sites in the human genome [8,9]. Most of these sites are in noncoding and repetitive elements regions, whereas RNA editing in coding regions is rare [10]. However, these editing events are very important because they can cause amino acid changes that lead to functional alterations of the encoded proteins.
These alterations may be responsible for activating oncogenes or inactivating tumor suppressors, thus affecting tumor progression. Several studies have reported a hypoediting pattern in glioblastoma (GBM) [11,12]. In GBM, unedited GluR-B enhanced malignancy [13], whereas A-to-I editing of AZIN1, which was increased in hepatocellular carcinoma specimens, resulted in amino acid changes that promoted cell proliferation [14]. We previously reported A-to-I editing of the Ras homologue family member RHOQ and demonstrated that edited RHOQ was associated with increased invasiveness [15]. These results show the importance of RNA editing in cancer, but few studies have investigated RNA editing sites in colorectal cancer (CRC).
In this study, we focused on finding nonsynonymous RNA editing sites in CRC using RNA sequencing data from The Cancer Genome Atlas (TCGA) database [2]. To accomplish this, we analyzed 39 paired colorectal tumors and adjacent normal tissue samples to identify recurrent editing sites that could be used as biomarkers for detection of CRC.
Materials and Methods
1. Nonsynonymous A-to-I RNA editing calling pipeline
We acquired RNA sequencing data from the TCGA database (https://tcga-data.nci.nih.gov) [2]. Bam files were analyzed using VarScan2 [16]. We compared data from normal and tumor samples with the following variant calling parameters: min-coverage, 8; min-var-freq, 0.2; mina-avg-qual, 20; and min-reads, 2. Variant call files were annotated with ANNOVAR [17]. We selected the positions that were converted from A to G, nonsynonymous single nucleotide variants (nsSNVs), after filtering somatic mutations that were enrolled in TCGA. We also removed all of the data from dbsnp137, excluding cDNA data and the positions within similar genomic regions.
2. Primary human colorectal tissues
To compare GLI1 RNA editing between tumor and normal tissue samples, 10 human colorectal tumor tissues and the matched normal tissues were obtained from the tissue bank of Seoul National University Hospital (Seoul, Korea).
3. DNA and RNA isolation and cDNA synthesis
Genomic DNA (gDNA) from cell lines was isolated using the QIAMP DNA mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Total RNA was extracted from each cell line using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions, after which 1 μg of total RNA was used to synthesize cDNA with random hexamers and ImProm-II reverse transcriptase (Promega, Madison, WI).
4. Polymerase chain reaction and Sanger sequencing analysis
RNA editing sites were amplified by polymerase chain reaction (PCR) using target-specific primers in gDNA or cDNA templates. Information regarding the primer is described in S1 Table. Purified PCR products were sequenced with a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Carlsbad, CA), then analyzed with a 3730 ABI capillary electrophoresis system (Applied Biosystems).
5. Pyro-sequencing analyses of GLI1 RNA editing
We designed GLI1 target-specific primers for pyrosequencing analysis. The reverse primer was biotin-labeled. Single-stranded biotinylated PCR products were processed for pyrosequencing analysis according to the manufacturer’s standard protocols (PyroMark Q96 ID, Qiagen). The primer sequences were as follows: forward, 5’-GACCGTCCTGCTCCAGCTAG-3’; reverse, 5’-CCCAACTTCTGGCTCTTCCTGT-3’; sequencing, 5’-AATGCTGCCATGGAT-3’.
6. Inosine chemical erasing assay
The inosine chemical erasing (ICE) assay is composed of four steps: (1) RNA cyanoethylation, (2) cDNA synthesis by reverse transcription, (3) PCR amplification, and (4) direct sequencing. We cyanoethylated total RNA from cancer cells containing edited transcripts for 15 minutes at 70°C alongside controls lacking cyanoethylation. The ICE assay was performed as previously described [18].
7. Cell culture and siRNA transfection
SNU-254 and SNU-81 human CRC cell lines were obtained from the Korea Cell Line Bank [19]. The cell line was grown in RPMI-1640 with 10% fetal bovine serum and gentamicin (10 μg/mL) at 37°C in a 5% CO2-humidified atmosphere. siRNA was purchased from Mbiotech Inc. (Seoul, Korea). A total of 20 nM siRNA was applied to cells in culture using G-fectin (Genolution, Seoul, Korea) according to manufacturer's standard protocols.
8. Western blotting
Cultured cells were washed with ice-cold phosphate-buffered saline and lysed with lysis buffer (50 mM Tris-HCl, pH 7.5, 1% NP-40, 0.1% sodium deoxycholate, 150 mM NaCl, 50 mM NaF, 1 mM sodium pyrophosphate, 1 mM EDTA, and protease/phosphatase inhibitors). Lysates were transferred to new eppendorf tubes after centrifugation at 13,000 rpm for 20 minutes. Protein concentrations were quantified with a Bicinchoninic Acid Protein Assay Reagent (Pierce, Rockford, IL) according to manufacturer's instructions. Samples containing equal quantities of total proteins were resolved on sodium dodecyl sulfate–polyacrylamide denaturing gel, then transferred to nitrocellulose membranes. The membranes were subsequently incubated in blocking solution containing 1% skim milk and 1% bovine serum albumin for 1 hour at room temperature, then probed overnight at 4°C with primary antibodies (anti–adenosine deaminase acting on RNA 1 [ADAR1], Abcam, Cambridg, MA; anti-actin, Santa Cruz Biotechnology, Santa Cruz, CA).
Results
1. The pattern of RNA editing in nonsynonymous editing sites
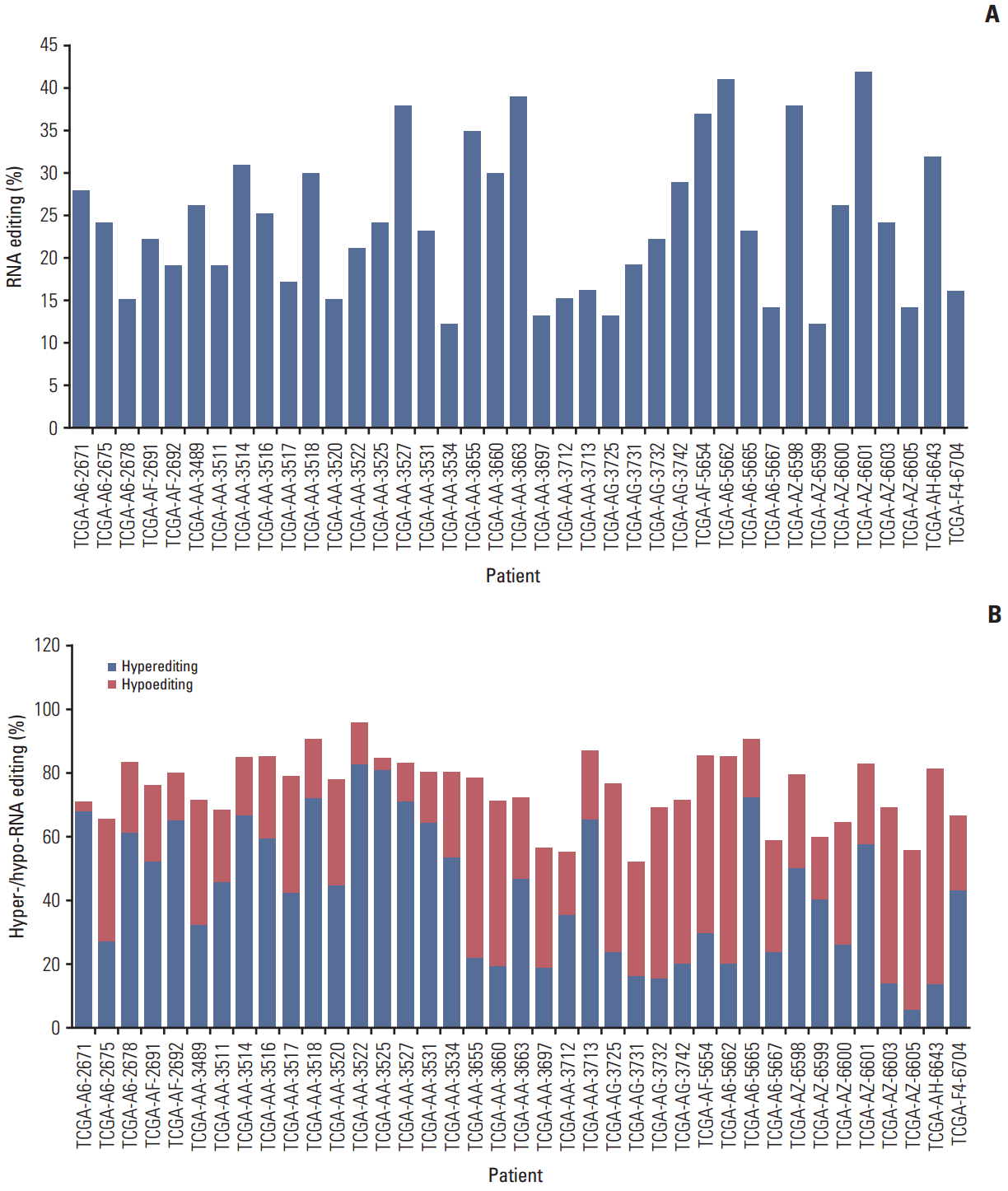
We downloaded 39 paired tumor and nontumor RNA transcriptome bam files from the TCGA database to analyze RNA editing patterns. The amount of total reads can influence the number of editing sites [20]; therefore, we evaluated only those samples with similar reads between normal and tumor tissues to reduce bias related to total reads. We utilized strict filtering conditions to find RNA editing candidates, using the VarScan2 software to identify RNA editing sites. VarScan2 was originally developed to identify somatic mutations and copy number alterations in exome sequencing data, but it can also be applied to analyze RNA-seq data. We set the analysis standards as follows: total reads, more than 8; editing frequency, 20; quality score, 20; and edited reads, 3. We then only selected nonsynonymous A to G conversions based on changes in AA in the refGene column, which was generated by ANNOVAR. We removed the positions enrolled in dbsnp137 except from those that originated from cDNA, similar genomic regions, and somatic mutations from the TCGA database (S2 Fig.). Overall, we identified 939 editing sites from 39 paired samples, including recurrent positions, ranging from 12 to 42 sites in each sample, with an average of 24 (Fig. 1A). We also investigated the percentage of hypo- and hyper-RNA editing within the samples. We defined hyperediting as a situation in which the percentage of RNA editing was 10% greater in tumors than adjacent normal tissue. There was no trend in hypoediting or hyperediting within the samples investigated. Among the 39 samples, 26 were classified as hyperedited and 13 were hypoedited (Fig. 1B).
We then focused on the recurrent hyper- or hypo-RNA editing sites in CRC. First, we listed the positions that were detected in more than two samples. The total number of recurrent detected editing positions was 39. Although we used relatively strict filtering standards, the possibility of false-positives remained; therefore, we employed additional filtering steps using the IGV AND PLAST programs. After these rounds of filtering, we found 10 recurrent editing positions in nine genes (Table 1).
2. The hypoediting sites in CRC
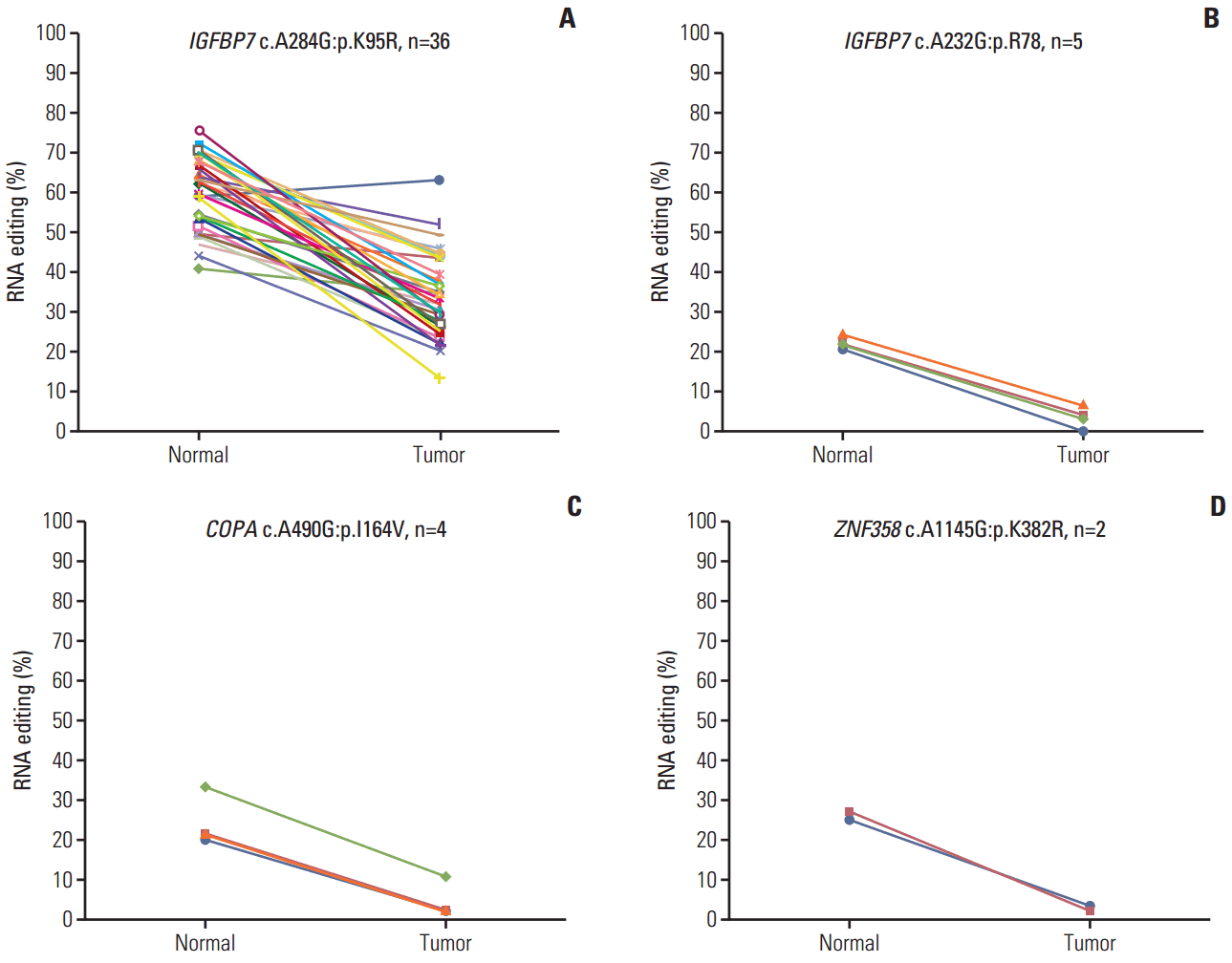
To identify RNA editing biomarkers for CRC, we investigated RNA editing by dividing edited messages into two categories: hypoediting, when the level of RNA editing is higher in normal than tumor samples; and hyperediting, when the level of RNA editing is higher in tumor samples. The results revealed four positions in three genes that were hypoedited in cancer. Hypo-RNA editing of IGFBP7 (c.A284G, p.K95R) was detected in 33 samples. The other hypo-RNA editing position of IGFBP7 (c.A232G, p.R78G) was detected in five samples (Fig. 2A and B), COPA, Coatomer Protein Complex, Subunit Alpha, was detected in four samples (Fig. 2C), and ZNF358, Zinc Finger Protein 358, was detected in two samples (Fig. 2D). These messages were significantly less frequently edited in cancer. Moreover, PDLIM, NEIL1, SRP9, and APMAP had both hyper- and hypo-RNA editing in 39 samples.
3. RNA editing of GLI1 is hyperedited in CRC
GLI1 RNA editing was detected in five samples, and was increased in colon cancer compared with normal tissue (Fig. 3A). We validated this position using Sanger sequencing (Fig. 3A) based on the pyrosequencing results of CRC cell lines (S3 Fig.). The guanosine peak was detected in cDNA, but not gDNA. After identifying GLI1 701G nsSNV, we confirmed the hyperedited pattern in CRC using Sanger sequencing in our tissue samples (Fig. 3C). There were increased guanosine peaks in tumor samples. To quantitatively analyze A-to-I RNA editing of GLI1, we conducted pyrosequencing in 10 paired samples of CRC and normal samples to measure the edited cDNA sequence. The extent of RNA editing of GLI1 was 29.3% in tumor tissues, while it was only 15.1% in normal tissues (Table 2, Fig. 3D). When compared with normal tissues, the level of RNA editing was increased by about 2-fold in tumor tissues. We also analyzed the clinical significance of GLI1 RNA editing in a TCGA cohort (n=39). More GLI1 was expressed in GLI1 RNA editing samples than in samples without GLI1 RNA editing (S4 Fig.). High expression or GLI1 RNA editing showed a significant relationship with the clinical prognosis (S5 Fig.). The clinical significance of GLI1 RNA editing merits further investigation in a larger patient population. Moreover, we found that the age, sex, primary site, histology, stage, microsatellite stable/microsatellite instable status, and KRAS mutation was not significantly related to GLI1 RNA editing (S6 Table). These data suggest that GLI1 is hyperedited in CRC.
4. Validation of the editing candidates
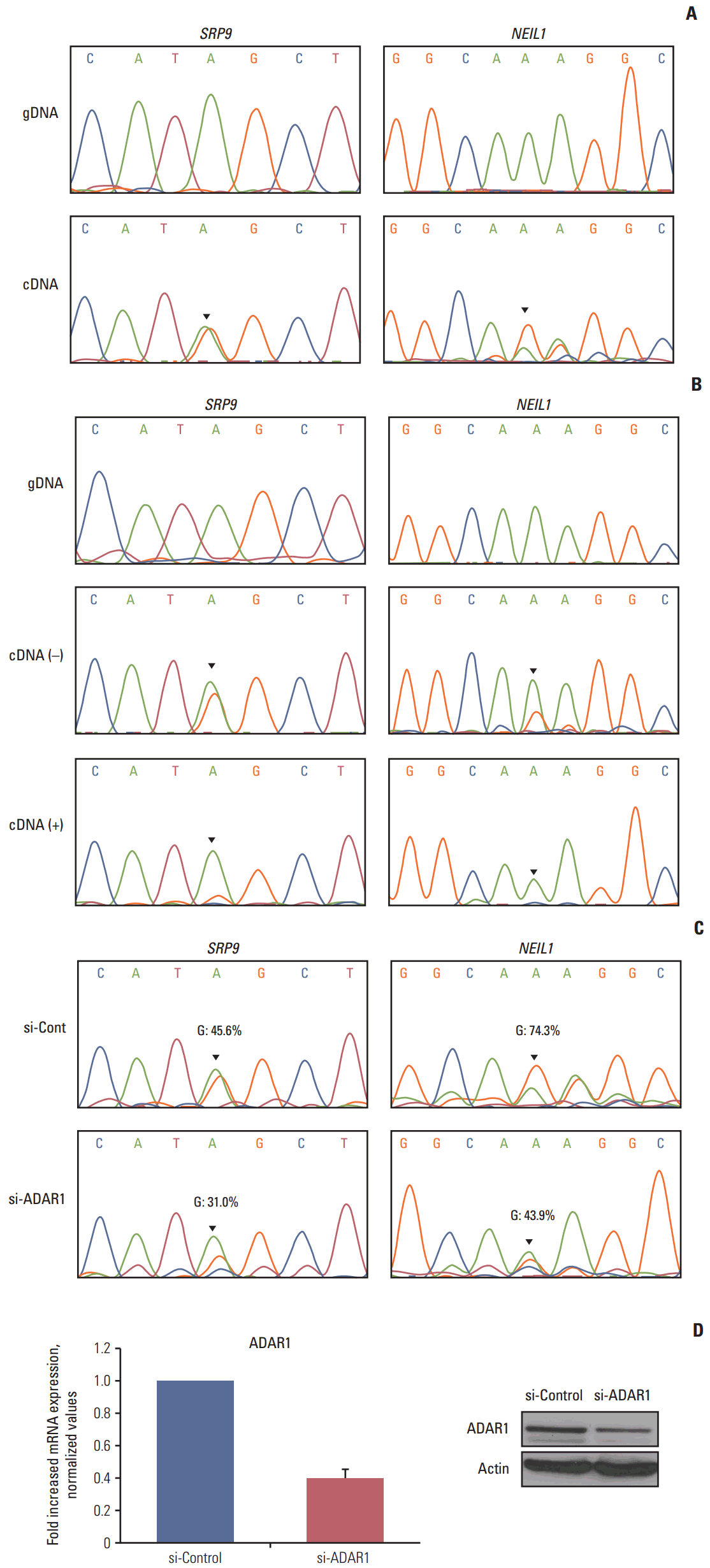
To confirm the reliability of our RNA editing discovery methods, we validated editing events using Sanger sequencing of gDNA and cDNA from the CRC cell lines. The cell lines that contain RNA editing were selected based on our RNA sequencing data. RNA editing of SRP9 and NEIL1 was validated because they were also detected in our cell line RNA sequencing data. Sanger sequencing confirmed the presence of a G peak in the cDNA of SRP9 and NEIL1 (Fig. 4A). The ICE assay is another method to validate A-to-I RNA editing, as cyanoethylated inosine will not be converted to guanosine by reverse transcriptase. We conducted an ICE assay to validate RNA editing of SRP9 and NEIL1. From these experiments, we found that the guanosine peaks were significantly decreased in the treated versus the nontreated samples in both positions (Fig. 4B). These data confirm the existence of A-to-I RNA editing from our analysis and, therefore, exclude the possibility of gDNA contamination.
ADAR family enzymes mediate A-to-I RNA editing. To investigate whether ADAR enzymes were involved in the RNA editing sites we discovered in CRC, we conducted RNAi-mediated ADAR knockdown. The expression of ADAR was reduced to approximately 40% of the wild type mRNA and protein levels (Fig. 4D). We next performed Sanger sequencing to determine the ratio of A/G. The editing level was reduced in both SRP9 and NEIL1 editing positions (G: 45.6%→31.0% in SRP9, G:74.3%→43.9% in NEIL1) in si-RNA ADAR treated cells (Fig. 4C). Although there were limitations to these samples, the results show that the identified RNA editing candidates exist, and are affected by ADAR enzyme knockdown.
Discussion
The development of high-throughput techniques has accelerated the identification of RNA editing events and broadened our insight into how the disease alters gene expression in ways other than genomic alterations. In higher eukaryotes, the most common type of RNA editing is adenosine to inosine [21]. A previous study has reported that other types of RNA editing are prone to false-positives [22]. RNA editing events in coding regions can result in amino acid changes that affect the progression of cancer; therefore; we focused on A-to-G RNA editing sites in coding regions.
In this study, we focused on nonsynonymous RNA editing in CRC. First, we investigated RNA editing patterns by comparing 39 paired normal and tumor tissues. The number of RNA editing sites ranged from 12 to 42 according to our analysis. Consistent with previous studies, our results showed that there were few editing events in coding regions. We also investigated hyperediting and hypoediting ratios per sample and found that there were different patterns between patients, and no consistent change between cancerous and normal tissue.
Although we used relatively strict filtering standards, the possibility of false-positives remained owing to the potential for mis-mapping or sequencing errors. We removed this possibility by filtering with the IGV and BLAST programs, after which 10 positions in nine genes with nonsynonymous alterations were identified at the RNA level. Because we did not obtain tissue samples that were enrolled in the TCGA dataset, we only validated two editing sites, which were also detected in our RNA sequencing data of CRC cell lines. The DNA repair enzyme, NEIL1 transcript, which contains RNA editing, has distinct enzymatic properties [23]. RNA editing of SRP9, which was first identified in breast cancer [24], was also identified in the present study. We validated these positions using Sanger sequencing and the ICE method. Furthermore, using siRNA, we found that the ADAR enzyme affects these editing sites.
We investigated normal- or tumor-specific RNA editing sites. Among the recurrently detected sites, four were edited less frequently in cancer. One of these is IGFBP7, which has been studied as a tumor suppressor gene in a variety of cancers [25,26]. Edited IGFBP7 transcript, which has editing sites A284G and A232G, is less prone to cleavage, indicating that hypoedited IGFBP7 transcript in cancer is likely to be cleaved, possibly affecting cancer progression. Hypoediting of COPA (I164V) has also been found in hepatocellular carcinoma [27], but the effects of editing have yet to be elucidated.
We also found that editing of GLI1 was increased in CRC. To validate this result, we investigated the level of RNA editing of GLI1 from normal and tumor tissue samples using pyrosequencing. These results, from an independent cohort, demonstrate that GLI1 was overedited in CRC. GLI1 editing is also found in medulloblastoma, but the extent of editing is decreased in the tumor [28]. These opposing result suggests an interesting hypothesis; in the medulloblastoma, Hedgehog (Hh) signaling, which involves GLI1, promotes cell survival [29]. However, GLI1, a transactivator of Hh signaling, inhibits cell proliferation in CRC cells [30]. Although identical editing events were generated, they could have different roles according to various tissue types.
We analyzed nonsynonymous RNA editing in CRC and found hypoediting or hyperediting sites. These results provide a comprehensive view of the editing pattern in CRC. RNA editing of GLI1 was found to be increased, suggesting that it could be a novel biomarker for CRC. In addition, understanding the different RNA editing patterns of GLI1 from different tissues and tumor types will enhance our understanding of the role of RNA editing in cancer.