Introduction
DNA is organized with histone and other proteins to form nucleosomes, which in turn are packaged into chromosomes. Histone acetylation is an epigenetic mechanism regulating chromosome configurations and gene expression. Two groups of enzymes control the acetylation status of histones: histone acetyltransferase (HAT) and histone deacetylase (HDAC) [1]. Since aberrant HDAC activity leads to transcriptional repression of tumor suppressor genes, HDAC inhibitors have been investigated for their anti-tumor effects [2]. Several preclinical studies found that HDAC inhibitors augment the effects of anti-cancer agents such as chemotherapy and ionizing radiation [2-4]. Numerous in vitro and animal experiments note that HDAC inhibition enhances radiosensitivity of diverse cancer cells [5]. Although HDAC inhibitors modulate radiosensitivity, the underlying mechanisms are not fully understood. HDAC inhibition may enhance radiation response by affecting cell functions such as gene expression, cell cycle and DNA damage repair.
In HDAC inhibitors-induced radiosensitization, temporal sequences between agents have a relevance to clinical practicability as well as mechanistic implications. In other words, clinical application of HDAC inhibitors as adjunct to radiotherapy should make the most use of a scheduling strategy that is logistically feasible and optimal for radiation enhancement. Most studies evaluated the effect of HDAC inhibition on radiosensitivity by exposing cells to HDAC inhibitors before irradiation. This strategy is likely to elicit maximum epigenetic modulation, and has generally proven the effectiveness of preirradiation treatment with HDAC to enhance radiation response [6-10]. However, some investigators opine that HDAC inhibition after irradiation is crucial to elicit optimal radiosensitization. Contrary to most studies, they found that preirradiation treatment with HDAC inhibitors has little effect on radiosensitivity, while significant radiosensitization is induced when cells are exposed both before and after irradiation [11-13]. Thus, the question regarding optimal combination scheduling of HDAC inhibitors and irradiation has not been yet answered.
The present study was conducted to investigate the effect of different sequences of HDAC inhibition and irradiation on radiosensitivity of human lung cancer cells. Cells were exposed to HDAC inhibitors trichostatin A (TSA) and SK-7041 before and after irradiation. We found that preirradiation TSA and SK-7041 treatment resulted in radiosensitization, while post-treatment showed much reduced effects.
Materials and Methods
1. Cell culture
A549 cell line was obtained from Korean Cell Line Bank (Seoul, Korea). Cells were grown as attached monolayers in RPMI 1640 media (JBI, Daegu, Korea) supplemented with 10% fetal bovine serum (JRH Biosciences, Lenexa, KS) and 12.5 µg/mL gentamicin (Gibco, Grand Island, NY). Cells were incubated at the exponential growth phase in humidified 5% CO2/95% air atmosphere at 37℃. Cells from the exponential phase were used for subsequent experiments.
2. HDAC inhibitors
TSA was purchased from Sigma Chemical Co. (St. Louis, MO). SK-7041 (4-dimethylamino-N-[4-(2-hydroxylcarbamoyl-vinyl) benzyl] benzamide 1), class I HDAC inhibitor previously reported [14], was a kind gift from Prof Yung-Jue Bang (Department of Internal Medicine, Seoul National University College of Medicine, Seoul). HDAC inhibitors were dissolved as concentrated stock solutions in dimethyl sulfoxide (DMSO), stored at -20℃ and diluted in culture medium before use. Control groups were treated with medium containing an equal concentration of DMSO.
3. Clonogenic assay
Details of the clonogenic assay methods were previously reported [8,9]. Cells were harvested from exponentially growing culture, and specified numbers were seeded into each well of six-well culture plates. Cells were treated with HDAC inhibitors for specified time, and the media was replaced by fresh inhibitor-free media before irradiation. Cultures were irradiated using 4-MV X-ray from a medical linear accelerator (Clinac 4/100, Varian Medical Systems, Palo Alto, CA) at a dose rate of 2.46 Gy/min. Cells were incubated for 14-21 days after irradiation till they were fixed with methanol and stained with 0.5% crystal violet. Colonies containing at least 50 cells were counted as clonogenic. Cell survival data were fitted to a linear-quadratic model using JMP software (SAS Institute Inc., Cary, NC). Surviving fractions were represented as a mean from triplicate experiments. Sensitizer enhancement ratio (SER) was defined as the ratio of radiation dose in the absence of HDAC inhibition to that in the presence of HDAC inhibition to produce a specified surviving fraction. Comparison of SER was done using paired t-test (Microsoft Excel 2010) between cells treated with HDAC inhibitors and untreated cells. Null hypotheses of no difference were rejected if p-values were less than 0.05. Type 1 error is not corrected for multiple comparisons.
4. Western blot for acetyl histone H3
Cells were washed, scraped and resuspended in cold lysis buffer (iNtRON Biotechnology, Seoul, Korea). The lysates were solubilized by sonication and centrifuged at 13,000 rpm for 20 minutes at 4℃, and the supernatant was collected. Equal amounts of protein were separated by 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA). Membranes were blocked with TBST blocking solution containing 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween 20 and 5% dry milk for 1 hour. Then, membranes were probed with polyclonal rabbit anti-acetyl-histone H3 immunoglobulin G (Upstate, Lake Placid, NY) at 1:2,000 dilution in 4℃ overnight, and washed and incubated with peroxidase-conjugated goat anti-rabbit immunoglobulin G (Jackson Immuno Research Laboratories, West Grove, PA) at 1:3,000 dilution for 1 hour. The same membranes were probed with monoclonal anti-α-tubulin antibody (Sigma) at 1:5,000 dilution at room temperature for 2 hours and incubated with peroxidase-conjugated goat anti-mouse immunoglobulin G (Jackson Immuno Research Laboratories) at 1:3,000 dilution for 1 hour. Antibody binding was detected using enhanced chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ). The optical density of each band was measured using Image J version 1.33u (National Institute of Health, Bethesda, MD). For quantitative analysis, density of acetylated histone H3 bands was standardized by that of corresponding α-tubulin bands.
5. Flow cytometric analysis
Cells were harvested using 0.25% trypsin and fixed in 1 mL of 80% ethanol (1-2×106 cells per sample). Cells were washed twice with phosphate buffered saline (PBS) and incubated in dark for 30 minutes at 37℃ in 1 mL of PBS containing 5 µg/mL propidium iodide (Molecular Probes, Eugene, OR) and 0.1% RNase A (Sigma). Flow cytometric analysis was done using FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ). At least 1×104 events were counted.
Results
1. HDAC inhibition before irradiation
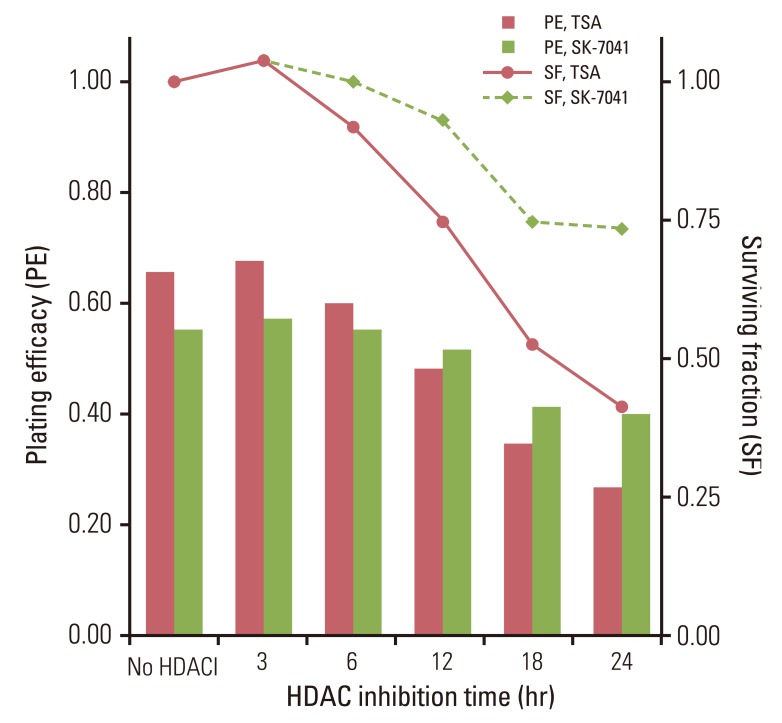
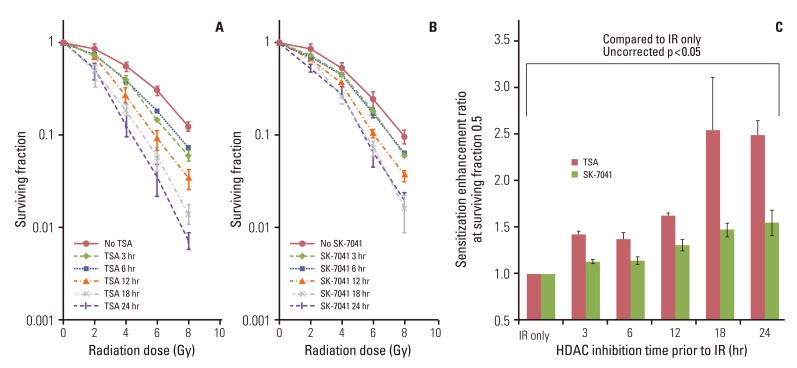
Treatment with HDAC inhibitors showed time-dependent cytotoxicity in A549 cells (Fig. 1). Both TSA and SK-7041 pretreatments enhanced radiosensitivity in A549 cells (Fig. 2A and B). Pretreatment-induced radiosensitization was closely associated with duration of HDAC inhibition. As A549 cells were treated for a longer time prior to irradiation, SER increased for both TSA and SK-7041 (Fig. 2C). SER of 18-hour treatment of TSA and SK-7041 were 1.68 and 1.60, respectively.
2. HDAC inhibition after irradiation
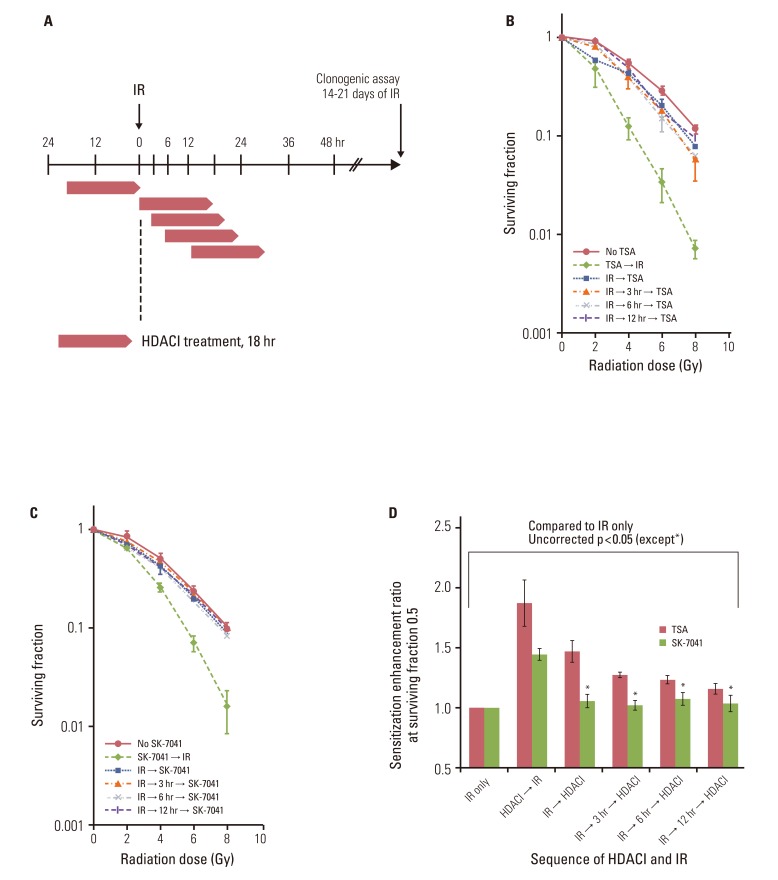
To investigate the effect of HDAC inhibition after irradiation, A549 cells underwent a series of combination sequences of HDAC inhibitor treatment and irradiation. HDAC inhibitors were added to culture medium immediately, at 3 hours, 6 hours, or 12 hours after exposure to X-rays, and clonogenic survival was determined (Fig. 3A). Compared with preirradiation TSA treatment, TSA post-treatment decreased the extent of radiosensitization (Fig. 3B). In contrast to SK-7041 treatment prior to irradiation, cell radiosensitivity was not apparently altered when A549 cells were exposed to SK-7041 immediately following irradiation or afterwards (Fig. 3C). Immediate postirradiation TSA and SK-7041 exposure induced radiosensitization by SER of 1.64 and 1.17, respectively. As treatment of HDAC inhibitors was delayed further following irradiation, their effect on cell radiosensitivity gradually diminished (Fig. 3D).
3. Histone H3 acetylation and irradiation
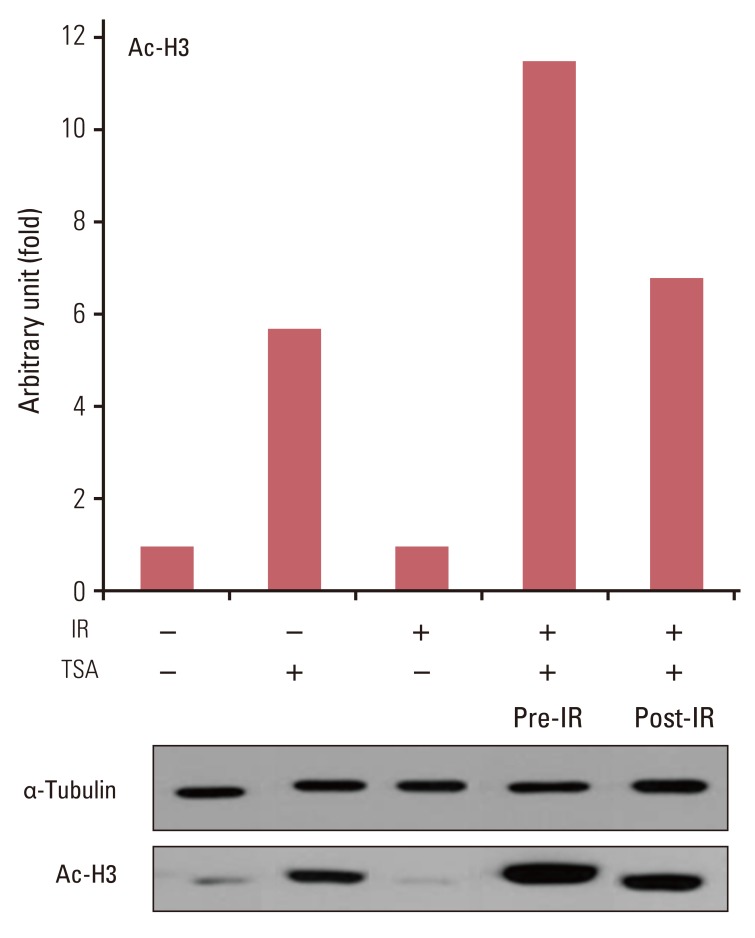
To address the mechanism through which HDAC inhibitors enhance radiation cell killing, analysis of acetyl histone H3 assessed the association of HDAC inhibition and the extent of radiosensitization. A549 cells were treated with TSA prior to or following irradiation and acetyl histone H3 was immunoblotted. Acetyl histone H3 increased by 5.7-fold in cells exposed to TSA compared to in untreated cells (Fig. 4). X-ray by itself had no effect on the level of acetyl histone H3. In contrast to irradiation of untreated cells, irradiation of TSA-treated cells induced additional 2-fold increase in acetyl histone H3 compared with TSA treatment without irradiation. When the sequence was reversed, postirradiation TSA treatment induced 6.7-fold increase in the level of histone H3 acetylation, comparable to that by TSA treatment only (5.7-fold increase).
4. Cell cycle profile following HDAC inhibition and irradiation
To investigate the effect of postirradiation HDAC inhibition on cell cycle progression, flow cytometric analysis was used to evaluate cell cycle distribution in A549 cells treated with various HDAC inhibition sequences. Flow cytometry revealed that either X-ray or TSA treatment arrested cell cycles at the G2/M phase (Fig. 5A). Postirradiation as well as preirradiation TSA treatment arrested cells in the G2/M phase. To distinguish the effects of radiation and HDAC inhibition, temporal changes in postirradiation G2/M phase proportion were analyzed. In TSA-naïve cells, the proportion of G2/M phase peaked (71.7% at 12 hours) after irradiation and rapidly plummeted below the baseline level at 36 hours (Fig. 5B). Preirradiation TSA treatment blocked irradiation-induced G2/M phase arrest. Immediate postirradiation TSA treatment had no effect on the peak extent of G2/M phase following irradiation. Delayed postirradiation TSA treatment (from 12 hours to 30 hours after irradiation) showed no apparent effect on the time course in G2/M phase proportion.
Discussion
Although the radiosensitizing capacity of HDAC inhibitors has been documented by preclinical studies [6-13,15], the optimal sequence of HDAC inhibition and radiation has not been fully defined. This study showed that preirradiation HDAC inhibition is essential for sensitization to radiation. Radiosensitization was directly commensurate with duration of HDAC inhibition prior to irradiation. These findings have potentially important implications for clinical application of HDAC inhibitors as radiosensitizers. Suboptimal scheduling in the clinical setting might fail to elicit the full effect of radiosensitization by HDAC inhibitors. Several preclinical studies reported that HDAC inhibitors enhanced radiation response. The majority of these studies found that preirradiation HDAC inhibition effectively induced a sensitization response [6-10,15]. Zhang et al. [16] reported that FK228 treatment prior to irradiation augmented cell radiosensitivity, while FK228 had no effect if cells were exposed to HDAC inhibitor following radiation. Combined with the present data, their findings support preirradiation HDAC inhibition as essential to elicit sensitization. For any given HDAC inhibitors, it is crucial that HDAC inhibition should be temporally coordinated with radiation to achieve optimal synergy.
Only a few researchers directly investigated the optimal sequence of HDAC inhibition and radiation [11-13]. Chinnaiyan et al. [13] reported that valproic acid induced comparable radiosensitization irrespective of sequences of HDAC inhibition and irradiation. The most effective scheduling to induce radiation enhancement is that cells are subjected to HDAC inhibition both before and after irradiation [13]. Other studies from the same group, using MS-272 and valproic acid, corroborated the argument that continued HDAC inhibition after irradiation is essential for maximal radiosensitization [11,12]. Given the diverse biologic effects of HDAC inhibitors, multiple mechanisms are implicated in enhancing radiation response. HDAC inhibitors epigenetically change gene expression profiles [1], which might render cells more susceptible to radiation killing. Based on the observation that delayed valproic acid treatment induces radiosensitization as effectively as preirradiation exposure, HDAC inhibition might interfere with late phases of DNA repair process by modulating chromatin remodeling [13]. Van Nifterik et al. [17] recently reported that postirradiation valproic acid treatment had no effect on radiosensitivity in human glioma cells, while preirradiation valproic acid exposure effectively augmented radiation lethality. Their findings contradict Chinnaiyan et al. [13]. Using the same HDAC inhibitor (valproic acid) and similar in vitro model (human glioma cells), two groups reported conflicting results with regard to optimal scheduling of valproic acid with irradiation. The present findings clearly showed that postirradiation HDAC inhibition by TSA and SK-7041 had little effect on cell radiosensitivity. Only pre-irradiation treatment of TSA or SK-7041 induces radiosensitization, which corroborates well the findings of Van Nifterik et al. [17] and Zhang et al [16]. These conflicting results might reflect a complex interplay between various HDAC inhibitors and different cell types in inducing radiosensitization. Existing evidence suggests that the relative contribution of preirradiation and postirradiation HDAC inhibition might differ among cell types and inhibitors. Thus, omission of preirradiation HDAC inhibitor treatment might risk suboptimal sensitization, until proven otherwise.
Several studies found a close association of HDAC and/or HAT with early responding components of DNA damage repair mechanisms. ATM [18] and 53BP1 [19] closely interact with HDAC early after irradiation. HAT is also closely related with DNA damage repair proteins. Sun et al. [20] demonstrated that ATM is rapidly acetylated through interaction with Tip60 HAT after DNA damage. They observed that the acetylation of ATM via Tip60 is a prerequisite for activation of downstream signal transducers, such as p53 and chk2. Although abundant literature exists regarding HDAC inhibitor-induced radiosensitization, research into HAT as a potential target for radiation response modulation has been limited. Suppression of Tip60 is associated with increased sensitivity to radiation lethality [20], suggesting a potential application of HAT inhibition as radiation response enhancer. Corroborating these observations, Sun et al. [21] reported that treatment with anacardic acid, which inhibits p300 and PCAF HAT, sensitizes tumor cells to radiation toxicity via inhibiting HAT activity of Tip60 and dependent activation of ATM and DNA-PKs. Existing research suggests that cellular sensitivity is augmented by pharmacologic inhibition of either of counteracting enzyme activity: HDAC [6-13,15-17] or HAT [20,21]. These counterintuitive results suggest that the nature of relationship of HDAC/HAT with radiosensitivity is far from straightforward. The present results also suggest that inhibition of HAT activity plays an important role in modulation of radiosensitivity. Irradiation induces the expression of acetylated histone H3 only in HDAC-suppressed cells (Fig. 4). In accord with Sun et al. [20], our findings imply that immediate early response to ionizing radiation is regulated by HAT. Radiation-induced histone H3 acetylation may be a manifestation of cellular radiation response driven by uninhibited HAT activity in HDAC-suppressed milieu. This view is corroborated by observations that that radiation-induced histone acetylation is lost unless irradiation is preceded by HDAC inhibition. The temporal window for HDAC inhibition to enhance radiation lethality coincides with that for irradiation-triggered histone H3 acetylation. Thus, the interpretation of the present data might be that both HDAC and HAT are involved in immediate responses to ionizing radiation. HDAC inhibition might induce radiosensitivity by disrupting coordinated function of HDAC/HAT in the early phase of DNA damage response.
Peri-irradiation HDAC inhibition is closely associated with regulation of radiation-induced G2/M-phase arrest. G2/M-phase arrest is an early response that cells adopt following exposure to ionizing radiation. The arrest of eukaryotic cells in G2 phase after irradiation is a universal phenomenon regardless of p53 gene status, and is associated with suppression of cyclin B [22]. There seems a direct association between G2 delay and cellular susceptibility to ionizing radiation: the longer the G2 phase delay, the more resistant cells are. This has led to a speculation among researchers that modulation in G2/M checkpoint might be a tool to enhance DNA damage after irradiation. Studies have demonstrated that augmentation of radiation lethality follows the inhibition of G2/M phase delay in human cancer cells by various pharmacologic agents such as caffeine, staurosporine derivative [23], c-Met inhibitor [24], and HDAC inhibitors [8,15]. In the present investigation, radiation-induced G2/M-phase arrest was blocked by preirradiation HDAC inhibition, but was not affected by HDAC inhibitor exposure after irradiation (Fig. 5). These observations lead to the speculation that preirradiation HDAC inhibition is crucial for radiosensitization, corroborated by the findings that HDAC inhibitors treatment prior to irradiation augmented radiation lethality in an exposure time-dependent manner (Fig. 2).
Conclusion
The current results demonstrate that the capacity of HDAC inhibitors TSA and SK-7041 to enhance radiation response requires preirradiation HDAC inhibition. Mechanisms underlying HDAC inhibitor sensitization might involve perturbation of early radiation response by suppressing HDAC activity and rendering HATs uninhibited, thus disrupting coordinated HDAC/HAT function in DNA damage repair. Before HDAC inhibitors are used as adjuvant to radiotherapy, optimal scheduling of two agents should be established from preclinical studies. Until the underlying mechanism is comprehensively described, it seems sensible to include preirradiation HDAC inhibition for maximal radiosensitization.