Introduction
Active hormone exposure is common in our lives, for example, from cigarettes, automobile exhaust, and cosmetic products, and the majority of these compounds are xenoestrogens, such as nonylphenol (NP), zearalenone, and bisphenol A. Xenoestrogens demonstrate estrogenic efficacy in the body through competitively binding to estrogen receptors. Estrogens regulate their effects through two estrogen receptors (ERs), ERα and ERβ, which differentially activate two downstream pathways. The estrogen response is tissuespecific and determined by the local ratio of ERα and ERβ. Generally, ERs exert their effects through both genomic and nongenomic mechanisms. In the former pathway, ERs activate gene transcription through interaction with estrogen receptor response elements in DNA. In other cases, ERs modulate diverse cellular pathways independent of interactions with DNA. For example, ERα promotes cell proliferation and survival via phosphoinositide 3-kinase activation, while ERβ reportedly inhibits cell cycle progression through interactions with cyclin D1 [1]. Recent studies demonstrated a seven-transmembrane G protein–coupled receptor, GPR30, rapidly responds to estrogen through cellular signaling [2]. GPR30 is reported to mediate cellular estrogen functions, including traditional genomic signaling through transcriptional regulation and rapid non-genomic estrogen signaling. The study demonstrated that estrogen activates mitogenactivated protein kinase/ERK1/2 through transactivation of epidermal growth factor receptor [3]. Subsequently, GPR30 was found to mediate estrogen-induced apoptosis through upregulating Bcl-2 expression [4]. Moreover, it was reported that estrogen induces c-fos transcription through ERK1/2 activation via GPR30, promoting the progression of breast cancer [5]. Thus, GPR30 seems to participate in estrogenmediated tumorigenesis and tumor progression, making it a potentially efficient therapeutic target for cancer treatment.
Colorectal cancer, also known as colon cancer, is one of the most common malignant gastrointestinal cancers [6]. The differential incidence rate between men and women suggests a hormonal role in the development of this disease [7]. The role of estrogen and xenoestrogens in cancer development has been intensively studied over the past decades. Bisphenol A reportedly increases the proliferation of human breast cancer cell lines in vitro and induces oxidative stress [8]. In addition, neoplastic transformation is observed in human breast epithelial cells exposed to bisphenol A [9]. It was reported that NP increases tumor formation and growth by inhibiting lymphocyte proliferation and macrophage activation [10]. The study demonstrated that NP enhances the progression of prostate cancer by modulating proteins that regulate the cell cycle, apoptosis, and metastasis, such as cyclin D1, cyclin E, p21, bax, and cathepsin D [11]. However, the effects of NP on colon cancer remain elusive.
Previous studies revealed a close relationship between colon cancer and 17β-estradiol (E2), the most abundant and most potent estrogen in humans. E2 plays a protective role in the development of colon cancer through ERβ, which is highly expressed in normal colonic mucosa but dramatically reduced in colon cancer [12]. Estrogen replacement therapy is reported to reduce colon cancer risk by 30%-40% in postmenopausal women [13]. It has been suggested that E2 inhibits the initiation and development of colon cancer, which could explain the lower incidence rate of colon cancer in women at all ages compared to men.
In the present study, we report that NP promotes proliferation and induces apoptosis failure in colon cancer cells through increasing expression of GPR30, which subsequently activates ERK1/2 signaling. In addition, the GPR30 inhibitor G15 abrogated NP-induced proliferation and inhibited tumor growth in vivo. Our study reveals the pro-tumorigenic role of xenoestrogens in colon cancer and suggests that GPR30 antagonism could be a novel therapeutic strategy for colon cancer.
Materials and Methods
1. Animals
Twelve-week-old male nude BALB/c mice were purchased and housed at the animal research facility of Zunyi Medical College. Mice were acclimatized to laboratory conditions and provided access to food and water ad libitum. Mice were kept for 7 days before experiments and housed throughout the study in an environment of 50±10% relative humidity, 24±2°C, and a 12-hour light/dark cycle.
2. Cell culture and cell treatment
The human colon cancer cell line SW480 and colorectal adenocarcinoma cell line COLO205 were obtained from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM) and minimum essential medium alpha, respectively. Complete culture medium was supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin, and cells were cultured at 37°C in a humidified atmosphere with 5% CO2. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA).
For proliferation and immunocytochemistry assays, cells were divided into three groups: (1) control group, (2) E2 group (cells treated with 10–7, 10–6, or 10–5 M of E2 for 24 hours), (3) NP group (cells treated with 10–7, 10–6, or 10–5 M of NP for 24 hours). For studying NP’s mechanism, cells were divided into three groups: (1) control group, (2) NP group (cells treated with NP for 0, 24, 48, or 72 hours), and (3) NP+G15 (cells pre-treated with G15 for 90 minutes followed by changing to medium containing NP).
3. MTT assay
To assess cell proliferation, 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyl tetrazoliumbromide (MTT) was used. Briefly, cells were seeded in 96-well plates at a final concentration of 2×104 cells/well. At the end of treatment, the media was removed, and 20 μL MTT dye solution (5 mg/mL in phosphate buffered saline [PBS] buffer) was added per well and allowed to incubate at 37°C for 4 hours. Then, the supernatant was decanted and replaced with 150 μL dimethyl sulfoxide. After 15-minute incubation with gentle shaking at 37°C, absor-bance was measured at a wavelength of 490 nm using a microtiter plate reader (Tecan, Maennedorf, Switzerland).
4. Flow cytometry for apoptosis analysis
Flow cytometry was employed to detect apoptosis. After treatment, cells were collected with trypsin digestion solution, washed twice with PBS, and resuspended in 200 μL PBS. For apoptosis analysis, the Annexin V/Dead Cell Apoptosis Kit (Invitrogen) was utilized according to the manufacturer’s instructions. Briefly, 5 μL Annexin V-FITC and 5 μL propidium iodide were added to each well, and the cells were incubated in the dark for 15 minutes. Then, apoptosis was measured using a FACScan flow cytometer (Becton, CA) equipped with CellQuest Software (Becton Dickinson).
5. Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, washed with PBS, and permeabilized with 0.1% Triton X-100 for 5 minutes. Samples were then blocked for 1 hour with 5% bovine serum albumin in PBS. Then, cells were incubated with rabbit anti-GPR30 (Abcam, Cambridge, MA) primary antibody for 1 hour at room temperature. Specific secondary antibodies conjugated with green fluorescent protein fluorochrome (Invitrogen) were subsequently incubated with cells for 1 hour at room temperature. After PBS washes, slides were mounted. Images were captured using an optical microscope (DP73, Olympus Corp.).
6. RNA extraction, reverse transcription, and quantitative polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions, and each sample was reverse transcribed to cDNA using the PrimeScript RT reagent Kit with gDNA Eraser (Takara, Dalian, China). Gene expression of GPR30, proliferating cell nuclear antigen (PCNA), c-myc, and cyclin D1 was measured by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) using an Applied Biosystems 7500 Fast RealTime qRT-PCR machine. Polymerase chain reaction primers used for reverse transcription polymerase chain reaction were synthesized by Invitrogen and sequences are as follows: hPCNA216 forward primers, CGGATACCTTGGCGCTAGTA; hPCNA-216 reverse primers, TCACTCCGTCTTTTGCACAG; hc-myc-175 forward primers, GGACTTGTTGCGGAAACGAC; hc-myc-175 reverse primers, CTCAGCCAAGGTTGTGAGGT; hcyclinD1-113 forward primers, AGCTGTGCATCTACACCGAC; hcyclin-D1-113 reverse primers, GAAATCGTGCGGGGTCATTG; hGPR30 forward primers, CTCTTCCCCATCGGCTTTGT; hGPR30 reverse primers, TACAGGTCGGGGATGGTCAT; hGAPDH forward primers, CCTCCCGCTTCGCTCTCT; hGAPDH reverse primers, CCGTTGACTCCGACCTTCAC.
7. Western blotting
Whole cell extracts were prepared from tissue or cells using RIPA buffer, and protein concentrations were determined using a bicinchoninic acid assay. Equal aliquots of proteins were separated using 8%-10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Following electrophoresis, separated proteins were transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA) and blocked with 5% BSA for 1 hour at room temperature. Membranes were then incubated overnight at 4˚C with primary antibodies. Primary antibodies used are as follows: anti-GPR30 (ab39742) and anti-GAPDH (ab8245) was purchased from Abcam (Cambridge, UK), anti-ERK1/2 (#9194), and anti-ERK1/2 (#8544), PCNA (#13110), ani-phosphorylated protein kinase A (pPKA) (#5661), anti-PKA (#4782), c-myc (#5605), and cyclin D1 (#2978) were purchased from Santa Cruz Biotechnology (Boston, MA). Membranes were then washed with TBST buffer and incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour. Protein bands were visualized using the enhanced chemiluminescence detection system (GE ImageQuantLAS4000, Tokyo, Japan). Band intensity was quantified using ImageJ software.
8. Soft agar colony formation assay
The soft agar colony-forming assay was performed to assess the colony formation and invasion potential of cancer cells. Briefly, 24 hours after treatment, cells were mixed with DMEM containing 0.35% soft agar (Sigma-Aldrich, St. Louis, MO). Then, cells were seeded in 35-mm dishes coated with DMEM containing 0.6% soft agar. After 10 days, the number of colonies were counted, and images were captured using an optical microscope (DP73, Olympus Corp.).
9. Tumor xenografts in athymic nude mice
SW480 cells were suspended in PBS at a final concentration of 1×108 cells/mL. A volume of 0.1 mL of suspended cells was subcutaneously injected into a single side of the posterior flank of each mouse (n=5/group). Five days after cell injection, tumor-bearing mice were randomly divided into three groups (6 animals per group): (1) control (no treatment), (2) NP treatment (80 μmol/kg), or (3) NP+G15 treatment (mice were administered 200 μg/kg G15 2 days before sacrifice). Mice were intraperitoneally injected with corn oil at a dose of 10 mL/kg for 14 days. At the end of the treatment, animals were sacrificed. Briefly, mice were anaesthetized with isoflurane in a closed chamber followed by exposure to carbon dioxide for ten minutes for euthanasia, and tumors were photographed.
10. Histology
Isolated lung tissues were embedded in paraffin and cut into 4 μM sections using a rotary microtome (Leica, Mannheim, Germany). Haematoxylin and eosin staining was used to visualize tissue structure. Dried slices were soaked in xylene, dewaxed for 10 minutes, rehydrated, rinsed in hematoxylin for 5 minutes and washed again prior to differentiation with 1% hydrochloric acid alcohol. Then, slides were stained with 0.55 eosin for 30 seconds, dehydrated with gradient alcohol, and soaked in xylene three times. Finally, slides were mounted using neutral gum. For immunohistochemistry staining, slides were stained with antibodies against Ki67 (#9449, Cell Signaling Technologies, Boston, MA) before hematoxylin co-staining. Histopathological changes of tissues were analyzed using an optical microscope (DP73, Olympus Corp.).
11. Statistics
Data are presented as the mean±standard error of mean. Statistical analysis between multiple groups was performed using one-way analysis of variance (ANOVA) with Dunnett’s post-test. Student’s t test was employed to compare differences between the two groups. A two-sided value of p < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA).
Results
1. NP promotes cell proliferation and apoptosis failure in colon cancer cells
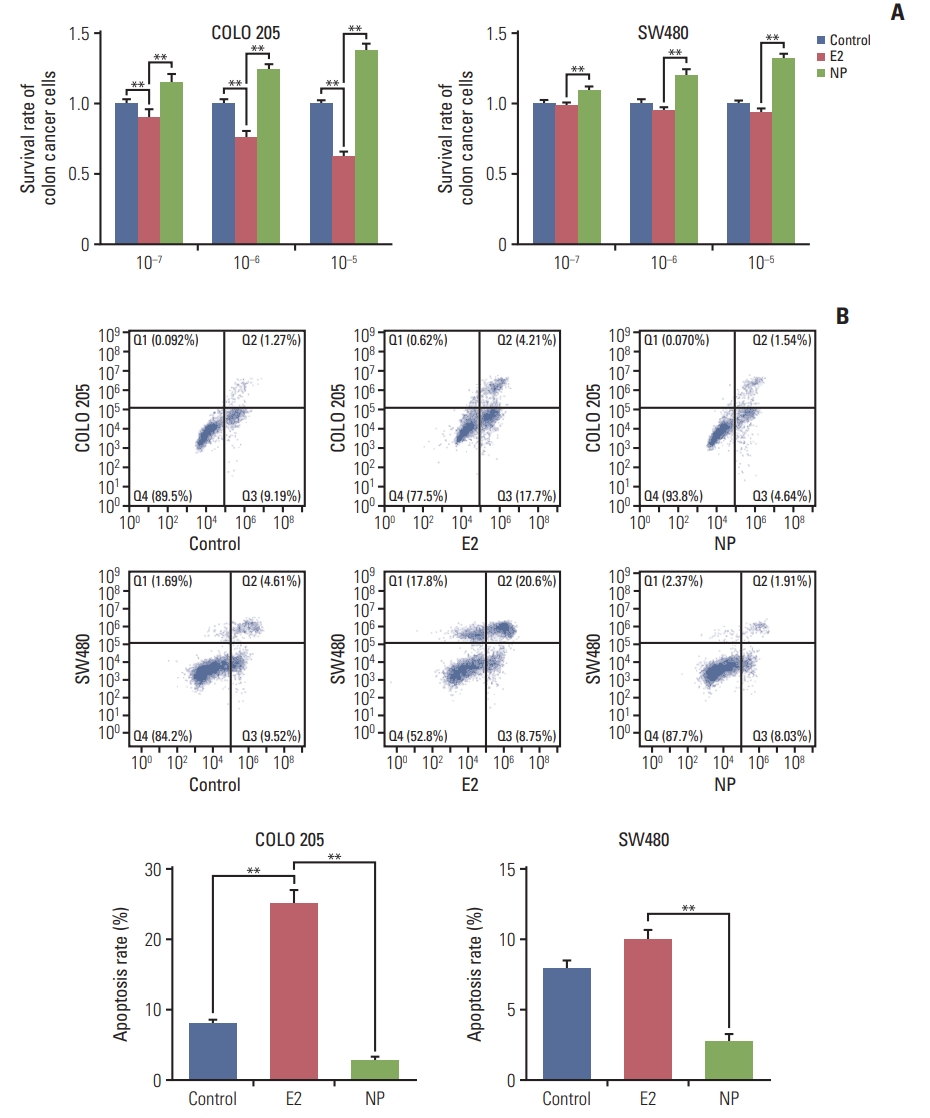
To investigate the effects of NP on colon carcinoma in vitro, the colon cancer cell line COLO 205 and the colon adenocarcinoma cell line SW480 were exposed to treatment with NP or E2, ranging from 10–7 to 10–5 μM. MTT assay revealed that E2 selectively reduced the proliferation rate of colon cancer cells, as cell viability was unchanged in SW480 cells treated with E2 (Fig. 1A). Meanwhile, NP dose-dependently increased cell viability of both cell lines. Next, flow cytometry was employed to assess apoptosis. As shown in Fig. 1B, E2 dramatically increased the rate of apoptosis in COLO 205 cells, but no significant alteration was observed in SW480 cells. However, NP significantly reduced the ratio of apoptotic cells. Taken together, these data suggest that NP facilitates cell proliferation and suppresses apoptosis in colon cancer cells.
2. NP-mediated cell proliferation is GPR30-dependent
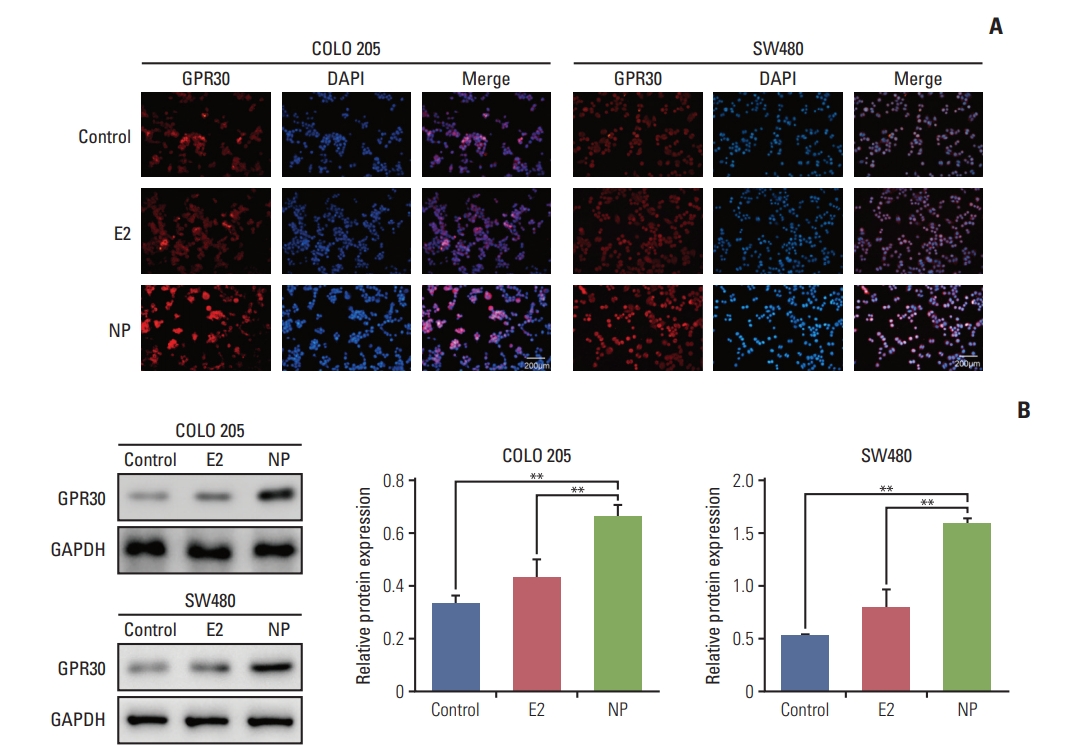
Environmental estrogens demonstrate relatively high binding affinities for the seven-transmembrane receptor GPR30 compared to classical estrogen receptors [14]. Furthermore, GPR30 has been implicated in regulating cell proliferation and invasion in numerous cancers [15,16]. Thus, we hypothesized that GPR30 participates in NP-mediated proliferation. Immunocytochemistry staining revealed that in COLO 205 and SW480 cells, no obvious changes occurred in GPR30 expression in the E2 treatment group (Fig. 2A). In contrast, NP significantly enhanced the expression level of GPR30, as shown by increased red fluorescence intensity of cells membranes. Next, total protein was extracted, and the expression level of GPR30 was assessed using Western blot. Consistent with the immunofluorescence results, Western blot demonstrated that NP increases the GPR30 expression level (Fig. 2B).
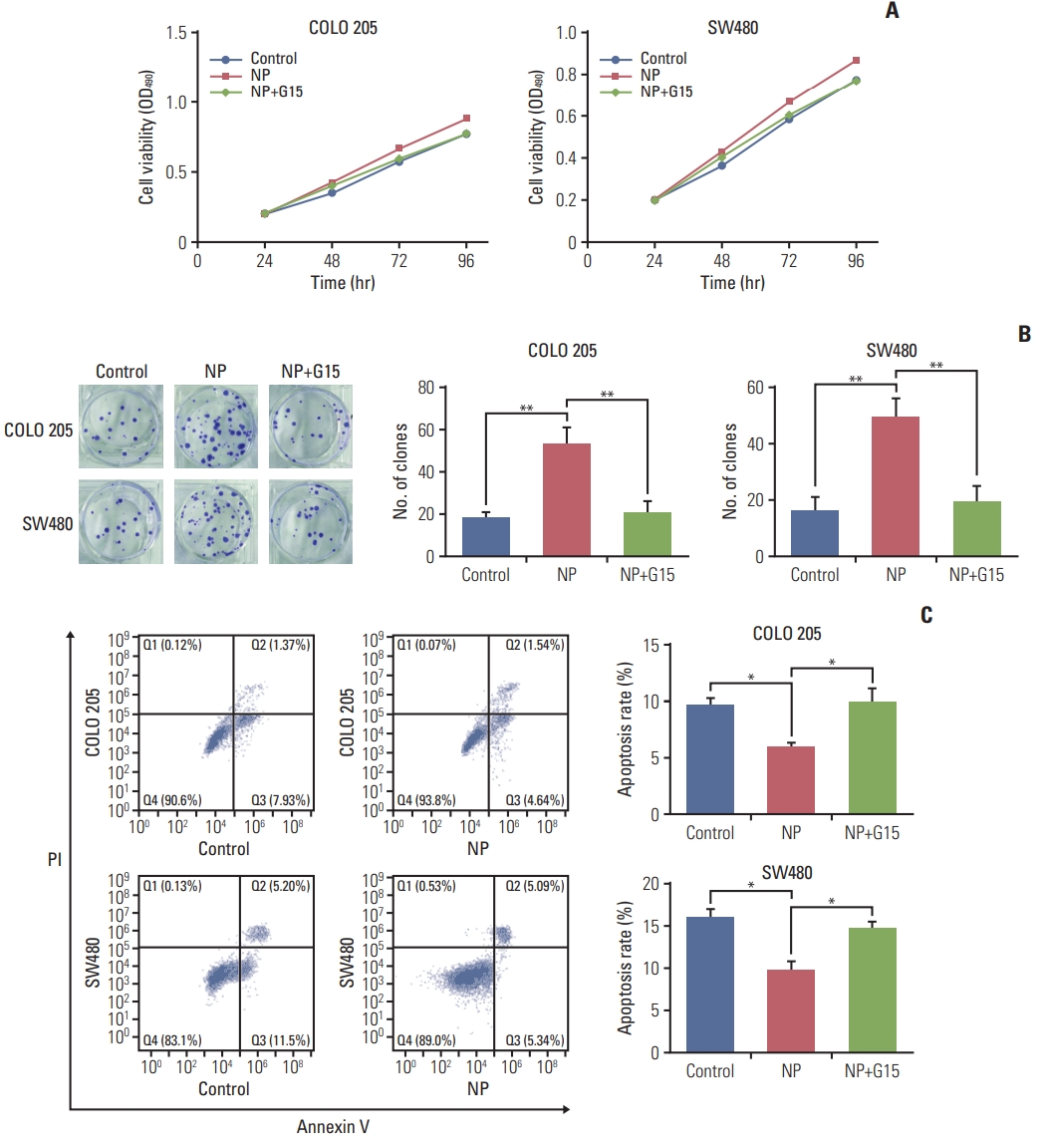
Next, G15, a GPR30 antagonist, was employed to confirm the role of GPR30 in colon cancer cells receiving NP treatment [17]. In COLO 205 cells, NP-induced cell proliferation was reversed by treatment with G15 (Fig. 3A), and similar results were observed in SW480 cells. Next, the colony formation efficiencies of COLO 205 and SW480 were evaluated using soft agar colony formation assay. NP significantly increased the colony formation capacity of colon cancer cells, which was dramatically inhibited in the G15 group. Moreover, NP-induced apoptosis failure was abrogated by G15 (Fig. 3C). Taken together, NP boosted the proliferation of colon cancer cells through activating GPR30, thereby protecting cancer cells from apoptosis.
3. NP regulates the proliferation of colon cancer cells through ERK1/2 signaling
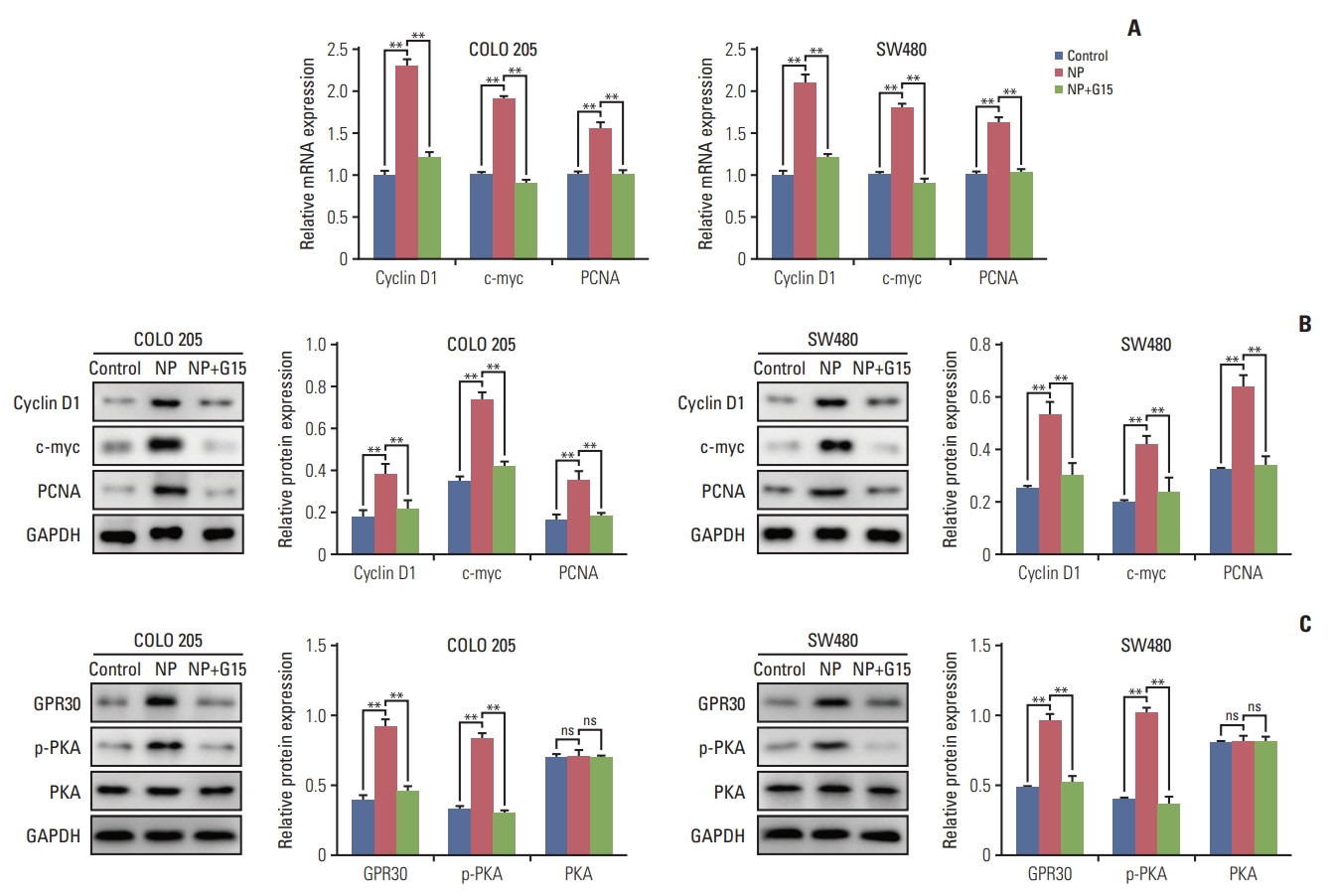
To unravel the mechanism by which NP-stimulated GPR30 regulates cell proliferation in colon cancer cells, we examined the effects of GPR30 on c-myc, a transcription factor that regulates tumorigenesis. mRNA levels of c-myc, proliferation marker PCNA, and cyclin D1 were significantly upregulated in response to NP treatment (Fig. 4A). Co-treatment of NP+G15 caused the NP-induced upregulation of the proteins mentioned above back to basal levels. The Western blot results were consistent with that of qRT-PCR (Fig. 4B). To assess the significance of GPR30 and p-PKA in NP-induced proliferation, we treated cells with either NP or NP+G15. The results showed that GPR30 and p-PKA increased in NP-challenged cells and decreased in NP+G15-treated cells when compared to the NP group.
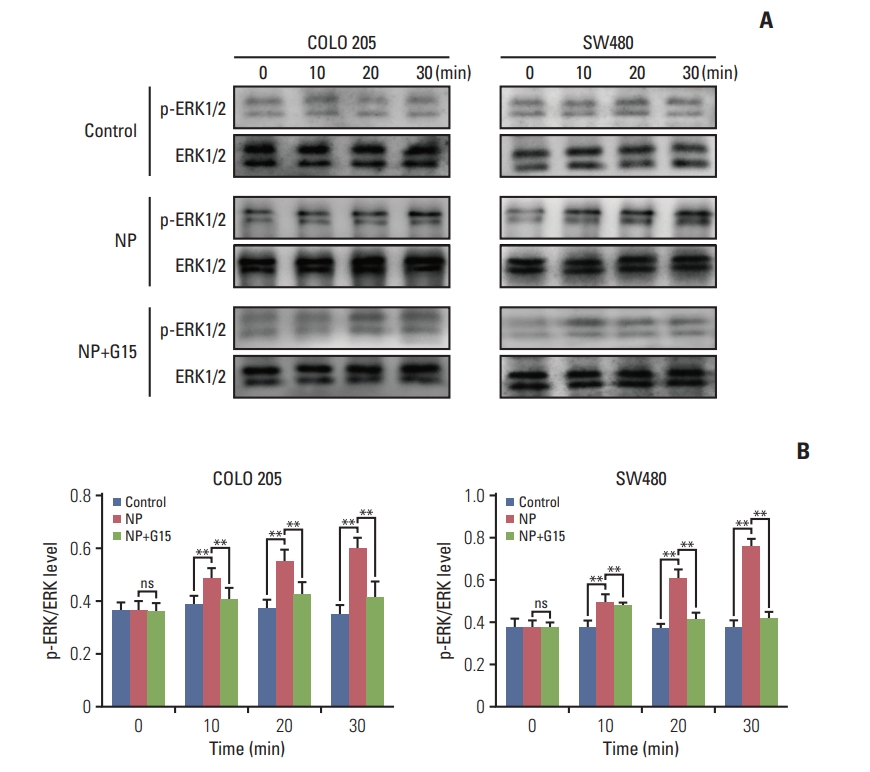
Previous studies reported that GPR30 promotes the growth of Sertoli TM4 cell through activation of ERK1/2 [18]. Next, we assessed the activation of ERK signaling and discovered that the ratio of p-ERK/ERK was remarkably increased by NP (Fig. 5A and B). Our results indicate that NP mediates the proliferation of colon cancer cells through the activation of ERK1/2 signaling.
4. NP facilitates tumor growth of colon carcinoma in vivo
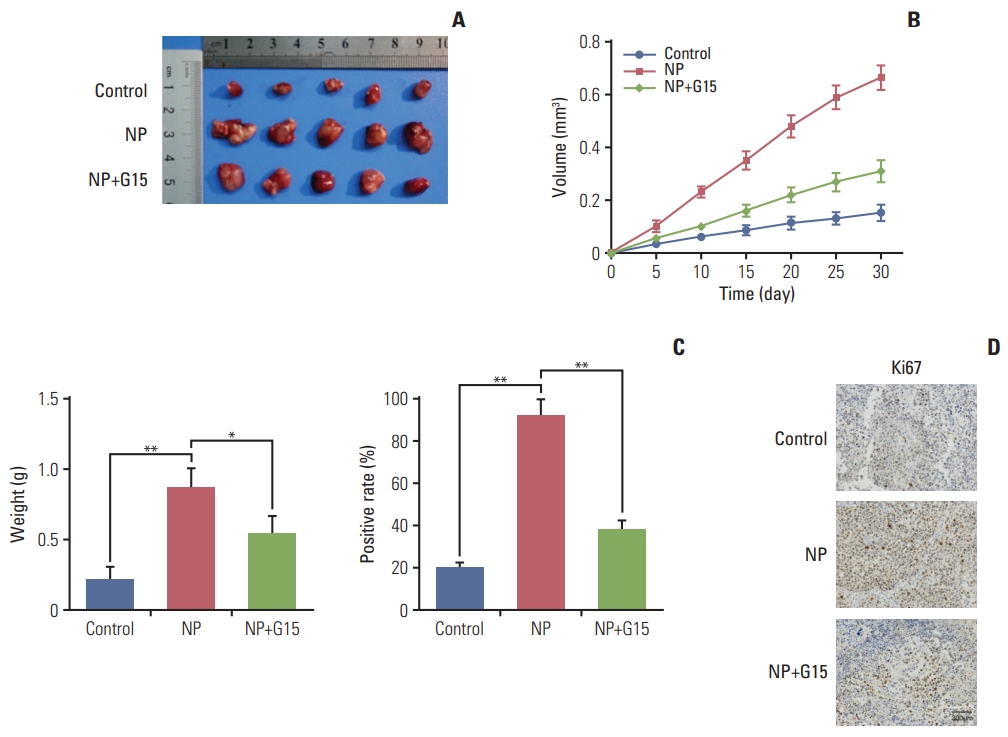
To evaluate the effects of NP in colon carcinoma tumorigenesis in vivo, COLO 205 cells were subcutaneously injected into nude mice. Mice were divided into control, NP treatment, and NP+G15 groups. At the end of 14-day treatments, animals were sacrificed, and tumors were excised. The results revealed that compared to the control group, the tumor size, volume, and weight were significantly inhibited by G15 treatment (Fig. 6A-C). Next, we investigated the expression of Ki67 in tumor tissues. Immunohistochemical data demonstrated that NP-induced increases in Ki67 were significantly suppressed by G15 (Fig. 6D). Taken together, these data suggest that NP promotes colon cancer cell proliferation in a GPR30-dependent manner.
Discussion
GPR30 is widely expressed in various tissues and cells, including lung, liver, prostate, and ovary. In addition, GPR30 is highly expressed in numerous estrogen relevant diseases and plays a role in E2-mediated efficacy, for example, in promoting cell proliferation, cell metastasis and enhancing apoptosis [2,14,19]. Arias-Pulido et al. [20] analyzed the expression of GPR30 in 88 primary inflammatory breast cancer patients and found that 69% of them were GPR30 positive. Co-expression of GRP30 and ER is associated with improved overall survival or disease-free survival. A significant increase in GPR30 expression was also reported in human non-small cell lung cancer cell lines compared to immortalized normal lung bronchial epithelial cells [21]. He et al. [22] reported that GPR30 mediates the invasion and carcinogenesis of endometrial cancer cell line RL95-2 through mitogen-activated protein kinase (MARK) activation. These findings suggest that GPR30 mediates the development of numerous tumors, and direct modulation of GPR30 might be a novel strategy for cancer treatment.
In the present study, we report that NP increases proliferation of COLO 205 and SW480 cells. Since ERα is absent in COLO 205 cells and endogenous ERβ was not detectable in SW480 cells, NP may exert its function independent of classic ERs [23]. Traditionally, the gene regulation effects of oestrogenic activities have been ascribed to two nuclear hormone receptors, ERα and ERβ [24]. However, over the past decades, a seven-transmembrane G protein–coupled receptor, GPR30, has been discovered as a novel mechanism of estrogenmediated rapid cellular signaling [2,25]. The affinity of E2 and GPR30 is 10 times greater than that of E2 for ERα, and GPR30 mediates E2-induced nongenomic signaling activities, including adenylyl cyclase stimulation, EGFR transaction, and MAPK activation [16]. In a study of 566 colorectal cancer patients, GPR30 expression was closely associated with poor survival in cancer stages 3-4 in female but not in male patients [26]. Herein, we also observed upregulated expression of GPR30 in colon cancer cells exposed to NP treatment, supporting the protumorigenic role of GPR30 in estrogen-mediated colon carcinoma proliferation and suggesting that intervention with GPR30 antagonists may serve as a novel therapeutic strategy.
NP has been shown to promote the proliferation of breast cancer cells as a consequence of the activation of ERα [27]. However, ERβ is the predominant estrogen receptor in colon cancer, with limited or no expression of ERα being detected. Expression of ERβ is down regulated in tissues of patients with colon cancer. Different from GPR30, ERβ usually conveys protective effects against colon cancer in vivo. NguyenVu et al. [28] elucidated that ERβ upregulates the expression level of miR-205, which targets oncogenic prospero homebox 1 via its 3′ untranslated region, suppressing the proliferation and metastatic capacity of colon cancer. Hartman et al. [29] demonstrated that ERβ inhibits colon cancer xenograft growth through cell-cycle modulation. Thus, the combination of ERβ and GPR30 antagonists would be a promising strategy for colon cancer control.
Taken together, our study indicates that NP affects apoptosis and proliferation of colon cancer through GPR30 and ERK1/2 signaling, suggesting that GPR30 is a potential target for colon cancer treatment.