Introduction
Pancreatic cancer, with a 5-year survival rate of less than 5% [1], is one of the deadliest known malignant tumors. Despite major advances in detection technologies and surgical techniques, the survival and prognosis of pancreatic cancer (PC) patients are still unsatisfactory. Presently, the curative treatment for PC is still surgical resection. For patients with ineffective surgical treatment or metastases, gemcitabine is still the first-line chemotherapeutic drug for PC [2]. As the etiology for this malignant digestive system tumor has not been fully elucidated, exploration of epigenetic factors that could be novel therapeutic targets or biomarkers of PC go mainstream [3].
As a third modification recently discovered in epigenetics, RNA post-transcriptional modification regulates RNA processing, stability, and metabolism [4]. RNA modification exists in almost all living organisms, recently, an increasing number of modifications, including 5-methylcytosine (m5C), N6-methyladenosine (m6A), and N1-methyladenosine (m1A), have been uncovered, among which m6A is the most prevalent internal chemical modification [5]. m6A is mediated by the m6A methyltransferase “writers,” eliminated by demethylases “erasers,” and recognized by binding protein “readers,” thereby influencing various biological processes. m6A RNA modification in PC has been confirmed. Guo et al. [6] verified that ALKBH5 activated PER1 by m6A demethylation and led to the reactivation of ATM-CHK2-P53/CDC25C signaling, which inhibited PC progression. Insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) serves as a reader for m6A-modified DANCR and stabilizes DANCR RNA to promote cancer stemness-like properties and pathogenesis [7]. Additionally, METTL14 overexpression in PC promotes cells proliferation and migration through targeting the downstream p53 effector related to peripheral myelin protein 22 (PERP) mRNA in an m6A-dependent manner, thus decreasing PERP expression [8]. Additionally, methyltransferase-like 3 (METTL3) knockdown in PC cells reveals enhanced sensitivity to the anticancer reagent gemcitabine [9].
Gemcitabine is a cell cycle-specific inhibitor and is the standard first-line chemotherapeutic drug for PC patients [10]. However, whether gemcitabine has a regulatory effect on m6A modification in PC is unclear. Here, we found that gemcitabine inhibited PC cell viability and reduced the overall level of m6A modification. Mechanistically, gemcitabine decreased the “writer” WTAP (Wilms’ tumor 1–associated protein) and m6A modification on MYC mRNA, thus decreasing its expression and interfering with the activation of the downstream pathways of MYC, inhibiting PC progression.
Materials and Methods
1. Collection of PC clinical information and samples
PC tissues along with adjacent normal samples were obtained from the Department of General Surgery, the First Affiliated Hospital of Soochow University. The overall experiment design and approach was approved by the Ethical Committee of the First Affiliated Hospital of Soochow University, and written informed consent was successively obtained from all participating patients before the study.
2. Cell culture
The human PC cell lines PANC1 and CFPAC1 (GenePharma, Shanghai, China) were cultured with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 1% antibiotics. The culture dishes were placed in an incubator at 37°C with 95% relative humidity and 5% CO2 partial pressure. The DMEM was changed every 2 days.
3. Transfection
Target genes small interference fragment and negative control fragment (si-NC) were purchased from Synbio Technologies (Suzhou, China). Cells were evenly plated and cultured to a density of about 70% and then transfected with 50 nM siRNA using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) to downregulate the target gene expression level according to the manufacturer’s instructions. After transfection, the cells were harvested for further experiments, and all the siRNA sequences are shown in S1 Table.
4. Cell viability assay
Cell viability was detected with a Cell Counting Kit-8 (CCK-8) kit (Dojindo, Kumamoto, Japan) according to the protocol. Approximately 5×103 PANC1 and CFPAC1 cells were seeded in 96-well plates per well after treated with or without gemcitabine for 48 hours, we checked relevant reports and conducted preliminary experiments, and finally set the gemcitabine concentration to 10 µM [11]. After incubated for 24, 48, 72, and 96hours, 10 µL CCK-8 was added and incubated at 37℃ for 2 hours, and the absorbance was detected at a wavelength of 450 nm.
5. 5-Ethynyl-2-deoxyuridine
A 5-ethynyl-2-deoxyuridine (EdU) analysis kit (RiboBio, Guangzhou, China) was applied to detect the cell proliferation capability. Stable transfected PC cells were incubated with diluted 5 µM EdU reagent for 3 hours. Then, cells were permeabilized and fixed for 30 minutes, then we used Apollo and DAPI regents to stain cell DNA and nuclei. Finally, EdU- and DAPI-positive images were taken with a fluorescence microscope.
6. Transwell migration assay
After treated with or without gemcitabine for 48 hours, appropriate 5×104 cells were evenly cultured in the upper chamber of a Transwell plate (Corning, Corning, NY) with serum-free DMEM, and 700 µL of DMEM containing 10% fetal bovine serum was added to the lower chamber. Then monitor cell growth status daily to ensure no apparent cells death. After normal culture for 48 hours, the cells were fixed with 4% paraformaldehyde and stained with crystal violet for 20 minutes, and the cells that migrated through the upper chamber membrane were counted. Images were acquired by inverted microscopy.
7. Quantification of overall m6A RNA methylation
We used EpiQuik m6A RNA Methylation Quantification Kit (colorimetric) (Epigentek, Farmingdale, NY) to quantify the overall m6A methylation level in PC cells. First, we extracted total RNA, and approximately 200 ng RNA was separated as an initial input. Then, the sample RNA, standard positive/negative control, and binding solution were added to the wells for 1 hour for assay and capture of the RNA according to the manufacturer’s instructions. After the samples were washed, the detection antibody and enhancer solution were added, and color developer solution and stop solution were added prior to measurement of the absorbance. The values were calculated using linear regression equations.
8. RNA extraction and real-time quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from PC cells or tissues with TRIzol for quantitative analysis. RNA was first reverse transcribed to cDNA with Vazyme reverse transcriptase at 42°C for 45 minutes following the manufacturer’s protocol, and PCR was then performed using SYBR Green qPCR Master Mix (GenePharma). Human ACTB was used as the housekeeping gene for mRNA expression. After 40 cycles of denaturation, annealing, and extension, the relative RNA levels were determined with the 2^-ΔΔCT method relative to the control. All the primers are listed in S1 Table.
9. Western blot analysis
A radio immunoprecipitation assay lysate mixture containing 1% phenylmethylsulfonyl fluoride was used to lyse and obtain total protein from PC cells. After quantification, electrophoresis was performed to separate the proteins, and then, the proteins were transferred to polyvinylidene difluoride (PVDF) membranes in transfer buffer. Then, the membranes were blocked in 5% skim milk at 37°C for 1 hour and incubated with primary antibodies overnight at 4℃. The next day, the membranes were incubated with an HRP-labeled secondary antibody for 1 hour at room temperature in the dark. Finally, immunoreactions were observed with a chemiluminescence detection system in a dark room.
10. WTAP and m6A immunoprecipitation assays
Immunoprecipitation assays of m6A and WTAP were performed with an RIP kit (BersinBio Biotech, Guangzhou, China). In brief, approximately 1×107 PC cells were collected and lysed for 30 minutes. DNA impurities were removed with DNase according to the protocol, and the supernatant was collected after centrifugation. The supernatant was then divided into two equal parts: one was incubated with anti-m6A or anti-WTAP primary antibodies overnight at 4℃ in a vertical orientation, and the other was used as input. Then the samples were incubated with target magnetic beads for 1 hour. After washing, RNA was extracted, and MYC expression was detected.
11. Immunofluorescence staining
To comprehensively observe the expression relationship between WTAP and the target gene MYC, immunofluorescence staining assay was conducted. First, after deparaffinization and antigen retrieval, the sections were fixed with 4% paraformaldehyde and blocked with 5% BSA (Solarbio, Beijing, China). Then, the sections were stained with WTAP primary antibody (Abcam, Cambridge, MA) and MYC primary antibody (Sigma, Darmstadt, Germany) at 4℃ overnight and incubated with the secondary antibody afterward. Finally, the cell nuclei were stained with DAPI, anti-quenching agent was added, and the sections were imaged with a confocal laser scanning microscope.
12. In vivo animal experiment
To evaluate the effect of gemcitabine on the tumor formation ability and gemcitabine/WTAP/MYC axis in vivo, we injected 2×106 PANC1 cells at logarithmic growth phase into the axilla of 4-week-old nude mice for subcutaneous tumor formation. The mice were randomly divided into phosphate buffered saline (PBS) group and gemcitabine treatment group. PBS or gemcitabine was injected into the mice via the abdominal cavity at a concentration of 50 mg/kg once every 5 days. The tumor dimension was measured every 5 days. After 30 days, the tumors were excised and weighed to compare the tumor size and volume, WTAP and MYC mRNA and protein expression was detected by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) and immunofluorescence. The care of laboratory animals was in accordance with the guidelines and ethical requirements of the Laboratory Animal Centre of Soochow University.
13. Statistical analysis
We used GraphPad 8.0 (GraphPad Software Inc., San Diego, CA) for data analysis and image generation. For statistical analysis, a two-tailed Student’s t test between two groups was performed. The Kaplan-Meier method for survival analysis. Correlational analysis of gene expression was conducted with linear regression. The scientific data are reported as the mean±standard deviation from three duplicate experiments. And the results were considered statistically significant when the p-value < 0.05.
Results
1. Gemcitabine inhibited PC cell proliferation and migration and decreased the overall m6A modification level
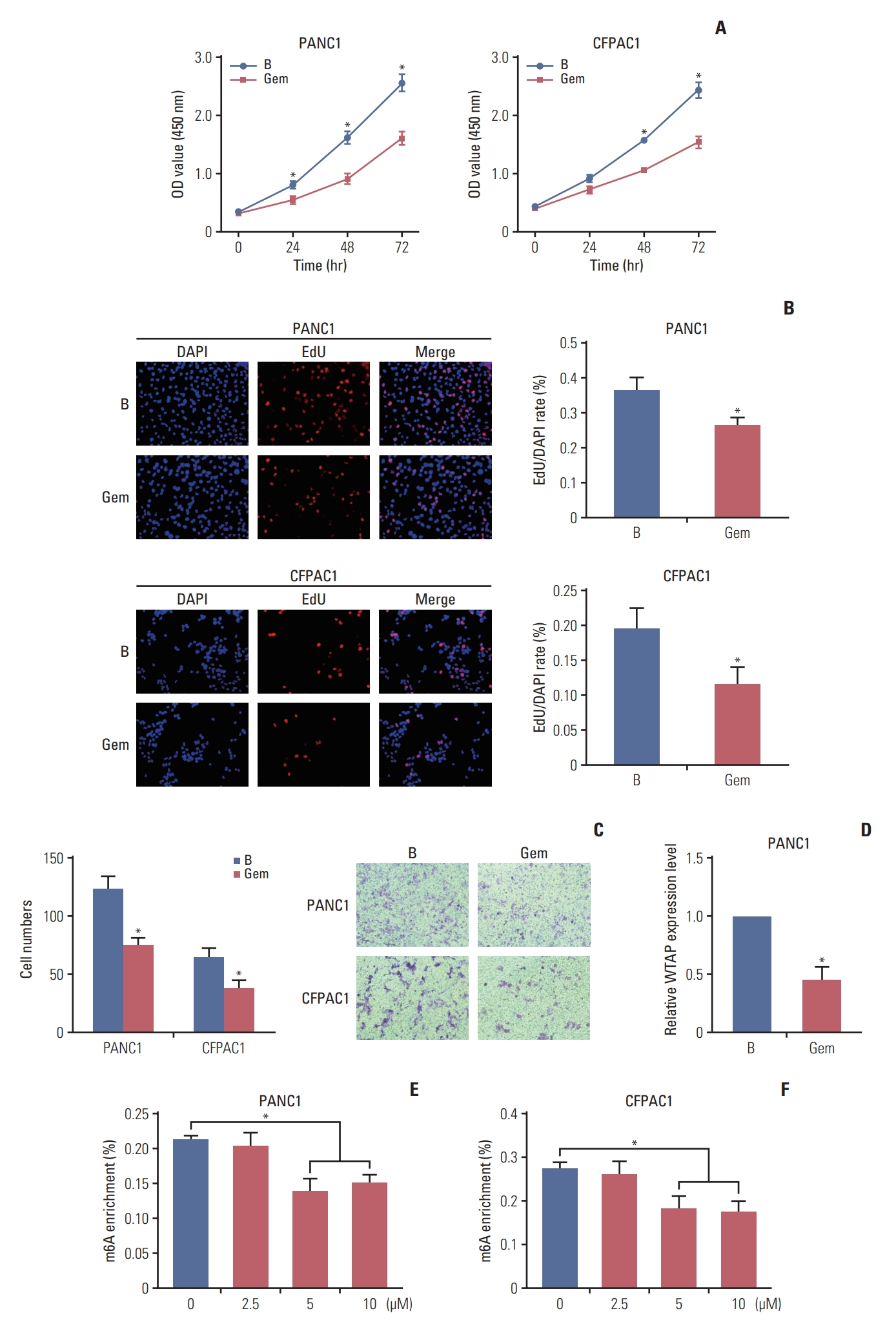
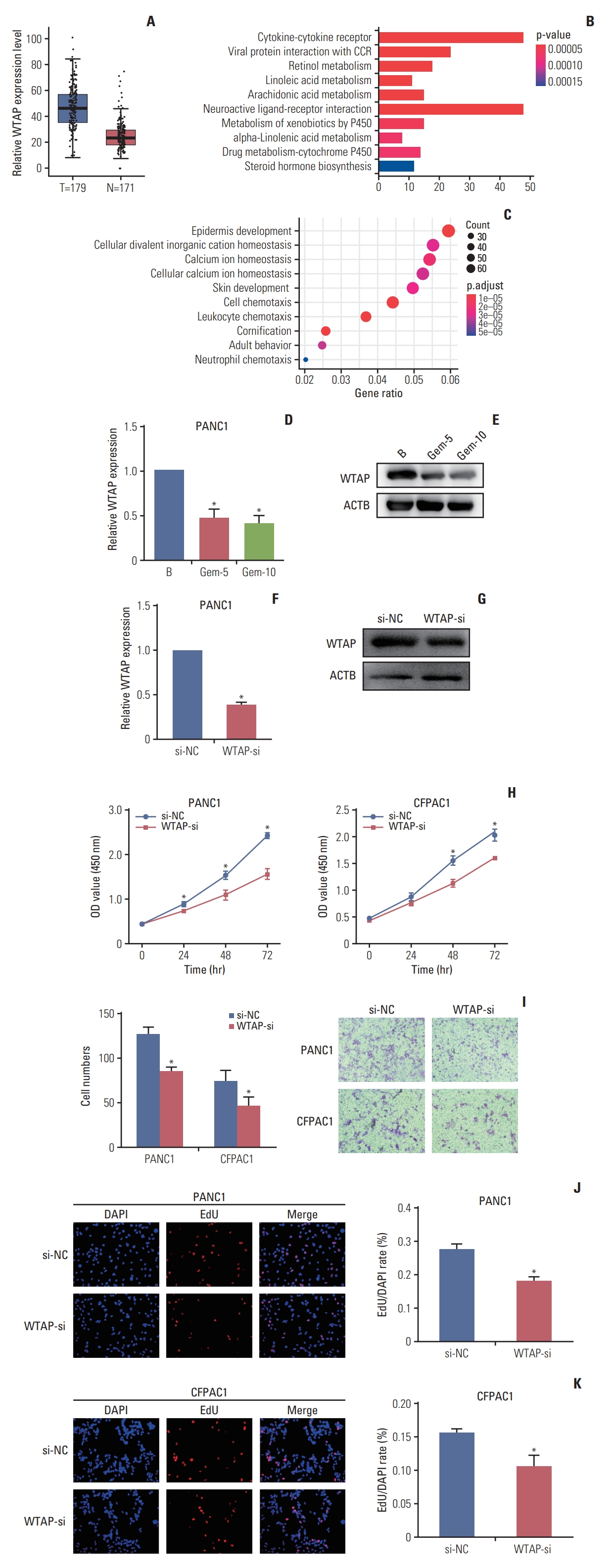
To explore the regulatory activity of gemcitabine on m6A RNA modification, cells were treated with gemcitabine to check the cell viability changes. CCK-8 and EdU assays revealed that gemcitabine inhibited cells viability (Fig. 1A and B). Transwell assays revealed the inhibition of cell migration ability by gemcitabine (Fig. 1C), revealing that gemcitabine was an effective treatment for PC. Subsequently, m6A quantification demonstrated that gemcitabine decreased the m6A modification level (Fig. 1E and F). Considering the inhibitory effect of gemcitabine on m6A level, we wanted to explore m6A regulators that were regulated by gemcitabine. qRT-PCR demonstrated that m6A writers and erasers were changed to varying degrees by gemcitabine, among which the writer WTAP was significantly decreased (fold change=0.37, p < 0.001) (Fig. 1D), suggesting that RNA methylation might be a target for gemcitabine. TCGA analysis revealed the similar upregulation of WTAP in PC (Fig. 2A). Taken together, we took WTAP as the target of gemcitabine for further research.
2. WTAP could be inhibited by gemcitabine and prevent PC cell proliferation and migration
Analysis of expression pattern of WTAP revealed its high expression in PC (Fig. 2A), we then divided 173 PC specimens into WTAP high and low expression groups according to the median WTAP level, Kyoto Encyclopedia of Genes and Genomes (KEGG), and gene ontology (GO) analysis revealed the important implications of WTAP on immunity and metabolism pathways (Fig. 2B and C). qRT-PCR and western blot analysis showed that WTAP expression level was reduced by gemcitabine and WTAP-si in PANC1 cells (Fig. 2D-G). CCK-8 and Transwell assays revealed that WTAP-si decreased cell viability and migration (Fig. 2H and I), which was also confirmed with EdU assays (Fig. 2J and K). Our results verified that important interaction between gemcitabine and WTAP might be significant for the clinical treatment of PC.
3. Gemcitabine decreased MYC stability by reducing the WTAP-dependent m6A-MYC modification level
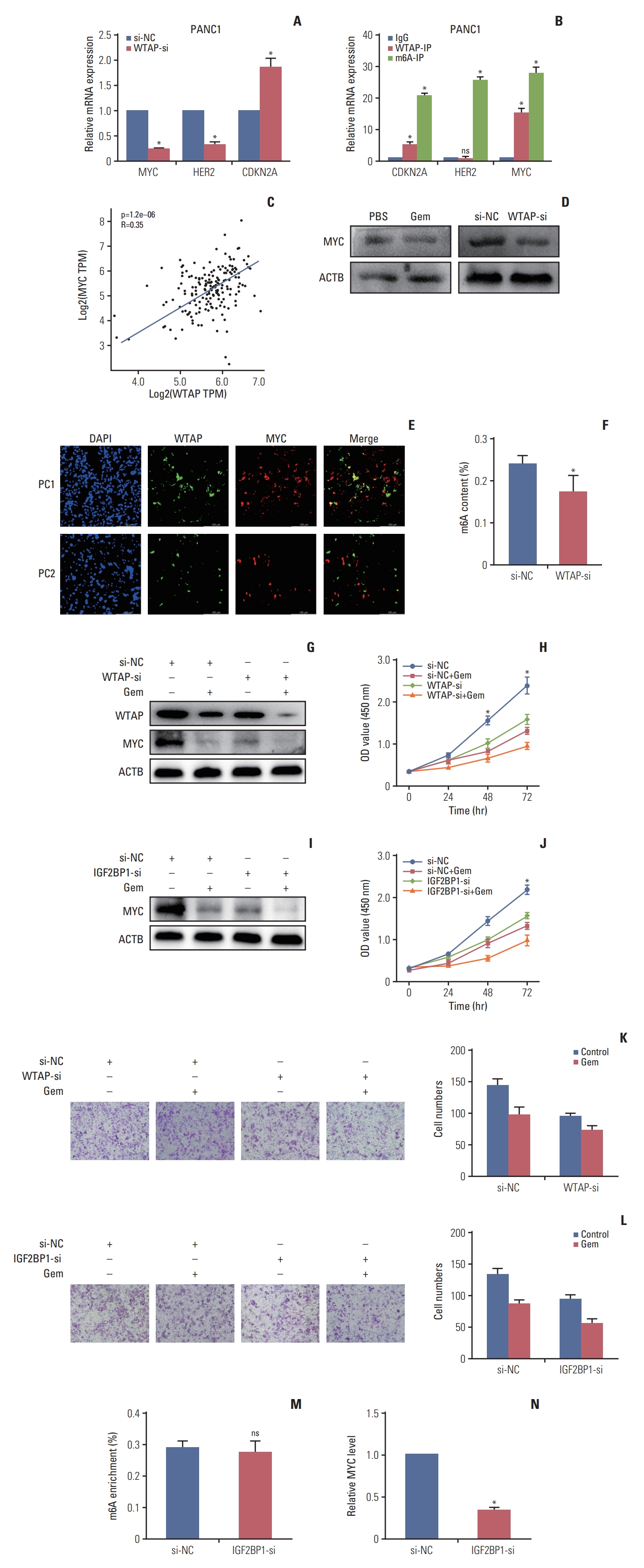
After identifying the regulation of gemcitabine on WTAP, we investigated which target was regulated by gemcitabine/WTAP. We first predicted possible downstream target genes of WTAP and detected their expression changes in WTAP knockdown cells. Experimental data showed that after WTAP knockdown in PANC1, MYC, and HER2 were downregulated with fold changes of 0.25 and 0.34, respectively, while CDKN2A was upregulated (Fig. 3A). WTAP-immunoprecipitation (IP) assay showed that CDKN2A and MYC, but not HER2, were significantly enriched, suggesting that CDKN2A and MYC might be WTAP targets. Then, m6A-IP revealed that CDKN2A, HER2, and MYC were all enriched in PANC1(Fig. 3B), this lead us to speculate that CDKN2A and MYC might be methylated by WTAP but HER2 might be methylated by other m6A regulators. TCGA analysis also preliminarily verified the positive correlation between WTAP and MYC (Fig. 3C). We knocked down WTAP with siRNA in PANC1 and observed decreased MYC protein expression (Fig. 3D). Immunofluorescence analysis of PC specimens also verified the co-expression relationship between WTAP and MYC (Fig. 3E). We further experimentally verified that the level of m6A modification was also inhibited after WTAP knockdown (Fig. 3F). To give an insight into the influence of Gem and WTAP on MYC, we treated WTAP-si PANC1 cells with or without Gem to monitor MYC expression verification, we verified Gem could restrain both WTAP and MYC expression in WTAP-si cells (Fig. 3G), CCK-8 and Transwell assay verified WTAP knockdown could enhance the inhibition of Gem on cell proliferation (Fig. 3H and K).
4. IGF2BP1 recognized m6A-MYC to regulate its stability and expression
The above results suggested that MYC is a crucial factor in PC progression. In addition, given that MYC is an indispensable proto-oncogene in various biological processes, such as metabolism [12] and the cell cycle [13], we believe that m6A-MYC is a novel treatment target in PC.
Studies found that IGF2BP1 could recognize m6A modification on MYC to alter MYC stability [14,15], so we speculated IGF2BP1 was a significant reader for m6A-MYC in PC. We found IGF2BP1 knockdown in PANC1 cells could also enhance the inhibition of Gem on MYC stability (Fig. 3I) and cell viability (Fig. 3J and L). What’s more, IGF2BP1 knockdown had no effect on m6A modification but inhibited MYC expression (Fig. 3M and N), Thus, we speculated that gemcitabine could interfere with WTAP-MYC-IGF2BP1 axis to inhibit PC progression.
5. Gemcitabine inhibited the PC cell tumorigenic ability in vivo by reducing the expression of the WTAP/MYC axis
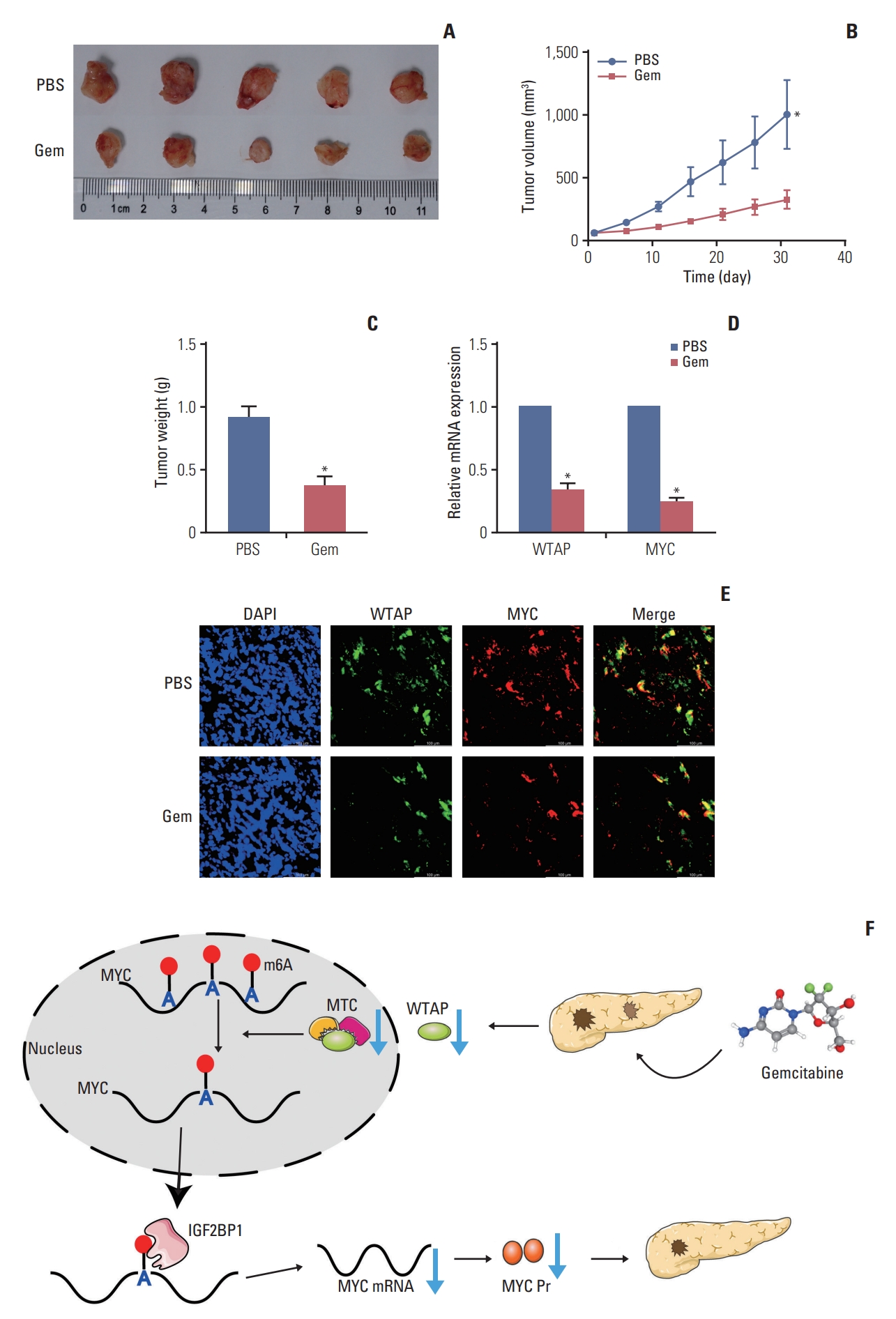
Next, we performed in vivo experiments to further verify gemcitabine on WTAP expression and the tumorigenic ability. Gemcitabine decreased the tumor volume, central necrosis rate, and tumor weight (Fig. 4A-C), WTAP and MYC also showed a downward trend after treated with gemcitabine (Fig. 4D and E). In conclusion, our experiments showed that gemcitabine inhibits WTAP expression and reduces the m6A modification on MYC and its stability, resulting in a decrease in MYC protein expression, thereby interfering with related pathways and inhibiting the progression of PC in vivo (Fig. 4F).
Discussion
PC, with unsatisfactory 5-year survival rate, is still one of the most aggressive diseases and has a poor survival rate. Surgery remains the most effective treatment for PC, even though over 80% of patients eventually develop local recurrence or metastases [16]. Various risk factors, including environmental and inherited factors [17], epigenetic chemical modification [18], type 2 diabetes mellitus [19], and pancreatitis [20], directly or indirectly contribute to the occurrence and development of PC. As PC is characterized by a strong interstitial hyperplastic reaction around cancer cells [21], its drug resistance and early invasive metastasis indicate the need for multidisciplinary comprehensive treatment, including surgery, chemotherapy and radiotherapy, immunotherapy and precision medicine and targeted therapy [22].
Increasing evidence has revealed that epigenetic deregulation is critically associated with tumor occurrence and pathophysiology, of which m6A RNA methylation is the most abundant epigenetic mechanism [23]. The m6A content is critical for cancer initiation, stem cell differentiation, metabolism, epithelial-mesenchymal transition, and drug resistance [23,24]. Research has shown that METTL3 interacts with the microprocessor protein DGCR8 to modulate miR-221/222 maturation in an m6A modification-dependent manner and promote bladder cancer proliferation [25]. Additionally, METTL14 could abolish the m6A modification of lncRNA XIST and recognition by YTHDF2 and augment XIST expression [26]. Another study showed that YTHDF1 was aberrantly upregulated in ovarian cancer and regulated EIF3C translation in an m6A-dependent manner, thus affecting overall protein translation in ovarian cancer [27].
As the first-line clinical chemotherapy for PC, gemcitabine can effectively inhibit the progression of PC and prolong the survival time of patients, and the disease control rate is up to 48.4% [28]. The related mechanism of gemcitabine in the treatment of PC was initially explored, but its relationship with m6A is poorly verified. We found that gemcitabine could decrease m6A methylation level in PC cells and inhibited WTAP expression, contributed to the reduction of m6A modification on MYC mRNA. This was a major change for MYC, which eventually led to a decrease in MYC protein expression and the inhibition of downstream pathways and ultimately limited the tumorigenic ability of PC cells.
As an oncogenic transcription factor, MYC can potentially regulate approximately 15% of genomic transcription [29]. MYC can regulate major downstream processes, including ribosome biogenesis, protein translation, cell cycle, and metabolism, thereby controlling cancer biological reactions, such as cell proliferation and differentiation [30].
However, our study still has some limitations, the effect of gemcitabine on other m6A regulators needs to be investigated. The regulatory relationship between gemcitabine and MYC downstream pathways needs to be deeply discussed. Moreover, whether MYC-related genes are methylated is also an important question and worth exploring. We did not expand the sample to verify the relationship between gemcitabine and m6A in PC specimens, and we will focus on clinical specimens in subsequent experiments.