Introduction
Pure red cell aplasia is a bone marrow failure characterized by a progressive normocytic anemia and reticulocytopenia without leucopenia and thrombocytopenia. It is associated with various diseases such as thymoma, lympho- and myelo-proliferative disorders, autoimmune diseases, infection, and drugs (1). Although various types of lymphoma may also be associated with this condition, the case of a pure red cell aplasia with angioimmunoblastic T cell lymphoma (AITL) has rarely been reported (2-6). We now describe a case of pure red cell aplasia associated with AITL. It was successfully treated with the aid of combination chemotherapy.
Case Report
A 43-year-old woman presented with malaise and fatigue that had developed one month prior. She was mentally retarded but was in good health until the symptoms developed. A CBC performed in a private clinic showed a hemoglobin level of 4.2 g/dL, a white blood cell count of 3,000/µL, and a platelet count of 234,000/µL. After a packed red cell transfusion, she was transferred to our hospital for further evaluation and treatment.
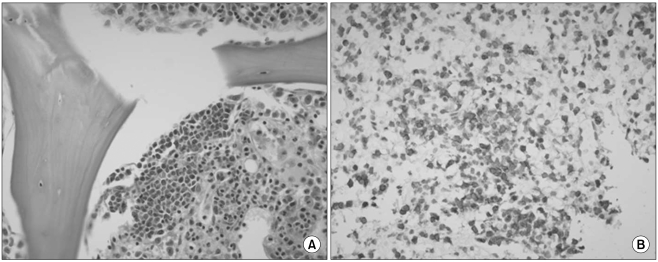
She had no signs of fever, weight loss, or night sweats. Laboratory finding at admission showed a hemoglobin level of 6.9 g/dL (MCV 84.9 fL), a white blood cell count of 3,900/µL, a platelet count of 197,000/µL, a reticulocyte count of 0.2%, serum ferritin of 814.8 µg/L, LDH of 664 U/L, total protein of 6.1 g/dL, albumin of 3.9 g/dL, and normal liver and kidney function tests. A peripheral blood smear revealed normocytic normochromic anemia. Direct and indirect Coombs' tests were indicative of strong positivity (4+). ANA was skeleton level 3, and C4 was decreased. A serologic test for EBV revealed that the patient was negative for EBV IgM and positive for EBV IgG. PCR for parvovirus B19 was negative. A CT scan of the abdomen revealed marked hepatosplenomegaly and small multiple lymphadenopathies in the aortocaval, para-aortic, and retrocaval areas. A PET-CT showed mild FDG uptake in the bilateral axillary, aortocaval, para-aortic, and retrocaval lymph nodes with hepatosplenomegaly and diffuse bone marrow involvement. A bone marrow biopsy revealed one lymphoid follicle, focal infiltration of abnormal lymphoid cells and the absence of red cell precursors (Fig. 1A). The infiltrated lymphoid cells were mostly positive for CD3. A chromosomal study showed 46, XX. These finding are consistent with a pure red cell aplasia with lymphoma involvement. A splenic biopsy showed diffuse infiltration of lymphoplasma cells admixed with congested vascular spaces. There were multifocal and perivascular aggregations of atypical lymphoid cells with clear cytoplasm, which were positive for CD3 (Fig. 1B). Despite the fact that there was not enough tissue biopsied to achieve a diagnosis, it was considered to be AITL. In accordance with the clinical manifestations, results of laboratory tests, and bone marrow and splenic biopsies, we finally diagnosed the patient as having a pure red cell aplasia associated with AITL (stage IVA).
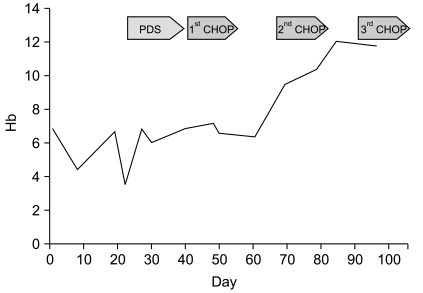
During diagnostic work-up, she developed fever, jaundice, hematuria, and skin rash on her whole body. Her hemoglobin rapidly decreased from 6.1 to 3.7 g/dL. Laboratory tests at that time were consistent with hemolytic anemia. She was treated with a washed RBC transfusion and prednisone but there was no response. The patient was started on CHOP (cyclophosphamide, adriamycin, vincristine, prednisone) chemotherapy. After one cycle, hemoglobin levels increased to 9.7 g/dL and all of the above noted symptoms disappeared. A bone marrow biopsy after one cycle of CHOP showed hypercellular marrow with erythroid hyperplasia. Fig. 2 describes change in hemoglobin levels during treatment. After 6 cycles of CHOP therapy, she achieved complete remission and a normal hemoglobin level and remained so out to 20 months.
Discussion
AITL is an aggressive mature T cell lymphoma. AITL patients present with systemic symptoms characterized by fever, skin rash, generalized lymphadenopathy, and hepatosplenomegaly. In addition, AITL has frequently been accompanied by various immunologic and hematologic diseases such as autoimmune hemolytic anemia, vasculitis, rheumatoid arthritis, and autoimmune thyroid disease (7). However, AITL associated with pure red cell aplasia is very rare.
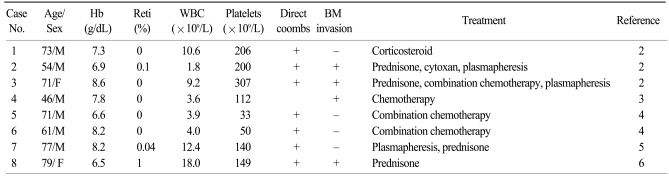
A MEDLINE search of the National Library of Medicine database (English literature only) yielded 8 additional cases of pure red cell aplasia associated with AITL. The main clinical findings of these patients are noted in Table 1. There were 6 male and 2 female patients, with a median age of 71 years (range: 46~79). The median hemoglobin level was 7.6 g/dL. All patients who had available laboratory data (7 patients) showed a positive direct Coombs' test. Bone marrow involvement in AITL was observed in 4 patients (50%). Although all patients were treated with corticosteroids, combination chemotherapy, and/or plasmapheresis, treatment outcomes were dismal. Five patients died of disease progression, infection, or pulmonary embolism. Corticosteroid and plasmapheresis resulted in only transient improvement. However, two patients who received combination chemotherapy achieved a complete response. In our case, prednisone alone did not improve the patient's hemoglobin level or reduce her systemic symptoms. However, after the 1st round of CHOP chemotherapy, hemoglobin levels increased and all of her previous symptoms disappeared. After 6 cycles of CHOP, she was in complete remission with a normal hemoglobin level. Therefore, aggressive combination chemotherapy should be used in pure red cell aplasia associated with AITL.
The pathogenesis of pure red cell aplasia associated with AITL has not been elucidated yet. Lynch et al. described significant inhibition of CFU-E cultures and modest inhibition of the BFU-E, but no inhibition of the CFU-GM from normal bone marrow using patient's serum (2). And a dose-dependent inhibitor of CFU-E was present in the serum of the patient (2). Other reports also demonstrated that the patient's serum before treatment significantly inhibited BFU-E formation but not CFU-GM from normal CD34+ progenitor cells (3). These results suggested that humoral factors causing inhibition of red cell precursors might play an important role in the pathogenesis of pure red cell aplasia associated with AITL.
The histological diagnosis of AITL is difficult and sometimes it has been misdiagnosed as other hematological or infectious diseases. In particular, the histological appearance of extranodal involvement such as bone marrow, spleen, skin and lung are usually nonspecific. In our case, we could obtain tissues from bone marrow and spleen but not lymph nodes because lymphadenopathies were existed only intraabdominal area. Although our patient did not express CD10 and EBV-LMP1, histological features and clinical findings were consistent with AITL.
In summary, pure red cell aplasia with AITL is a very rare condition and may be associated with an immune disorder. Although it has an aggressive clinical course and poor long-term outcome, the clinician must try to treat it with aggressive combination chemotherapy. Further investigation regarding the pathogenesis of and novel therapeutic approach to this rare condition, such as new immunomodulatory strategies, are needed to determine the optimal therapy of pure red cell aplasia with AITL.